






中国农业科学 ›› 2020, Vol. 53 ›› Issue (9): 1730-1742.doi: 10.3864/j.issn.0578-1752.2020.09.004
所属专题: 专题——限制性两阶段多位点全基因组关联分析法的应用
• 专题:限制性两阶段多位点全基因组关联分析法的应用 • 上一篇 下一篇
潘丽媛1,贺建波1( ),赵晋铭1,王吴彬1,邢光南1,喻德跃1,张小燕3,李春燕3,陈受宜2,盖钧镒1()
),赵晋铭1,王吴彬1,邢光南1,喻德跃1,张小燕3,李春燕3,陈受宜2,盖钧镒1()
收稿日期:2019-08-24
接受日期:2020-01-02
出版日期:2020-05-01
发布日期:2020-05-13
联系方式:
潘丽媛,E-mail:panly89@126.com。
基金资助:
LiYuan PAN1,JianBo HE1(),JinMing ZHAO1,WuBin WANG1,GuangNan XING1,DeYue YU1,XiaoYan ZHANG3,ChunYan LI3,ShouYi CHEN2,JunYi GAI1()
Received:2019-08-24
Accepted:2020-01-02
Published:2020-05-01
Online:2020-05-13
摘要:
【目的】为全面解析大豆重组自交系群体中调控百粒重性状的QTL体系,将限制性两阶段多位点全基因组关联分析方法(RTM-GWAS)和不同定位方法进行比较、优选,为后续候选基因体系探索及分子标记辅助育种设计提供依据。【方法】利用以科丰1号和南农1138-2为亲本衍生的重组自交系群体NJRIKY的427个家系,通过由全基因组39 353个SNP构建的3 683个SNPLDB标记及3个环境下的百粒重表型数据,选用复合区间作图法(CIM)、基于混合线性模型的全基因组关联分析方法(MLM-GWAS)和RTM-GWAS3种方法检测百粒重QTL,通过QTL数目和总的表型变异解释率比较检测功效,挑选最佳定位结果进行NJRIKY群体中的百粒重遗传体系解析。通过候选基因体系的功能注释,挖掘调控大豆百粒重的生物学途径。【结果】科丰1号与南农1138-2的百粒重差异较大,多环境平均数分别为9.0和17.9 g,遗传变异系数为12.4%,遗传率为85.4%,适用于百粒重性状的遗传解析。比较3种方法定位结果表明RTM-GWAS方法表现最佳,检测QTL数目最多(57个),解释表型变异最多(70.78%)。而CIM仅检测到14个QTL,解释了56.47%的表型变异,MLM-GWAS仅定位到6个QTL,解释了18.47%的表型变异。RTM-GWAS共检测到57个QTL,分布在19条染色体上,表型变异解释率为0.03%—7.57%,其中41个QTL覆盖了已报道的来自30个双亲群体的81个百粒重QTL,16个QTL为新发现位点,包含一个表型变异解释率大于3%的大效应位点Sw-09-2。此外,检测的57个QTL中有20个位点与环境存在互作效应。这57个QTL构成了影响NJRIKY群体百粒重性状的遗传体系。通过SNPLDB标记与预测基因内的SNP进行χ2检验,共筛选到36个候选基因,其中4个候选基因来自大效应QTL,剩余32个候选基因来自小效应QTL。通过GO注释发现这些候选基因功能注释丰富,其中13个候选基因与籽粒发育直接相关,剩余的候选基因功能丰富,包含转运、转录调节因子等,表明不同生物学途径的基因共同调控NJRIKY群体中百粒重性状的表达。【结论】3种定位方法中,高效的RTM-GWAS方法检测到较为全面的NJRIKY群体的百粒重QTL,更适用于双亲RIL群体的QTL定位。不同功能的候选基因共同调控了复杂的百粒重性状的表达。
潘丽媛,贺建波,赵晋铭,王吴彬,邢光南,喻德跃,张小燕,李春燕,陈受宜,盖钧镒. RTM-GWAS方法应用于大豆RIL群体百粒重QTL检测的功效[J]. 中国农业科学, 2020, 53(9): 1730-1742.
LiYuan PAN,JianBo HE,JinMing ZHAO,WuBin WANG,GuangNan XING,DeYue YU,XiaoYan ZHANG,ChunYan LI,ShouYi CHEN,JunYi GAI. Detection Power of RTM-GWAS Applied to 100-Seed Weight QTL Identification in a Recombinant Inbred Lines Population of Soybean[J]. Scientia Agricultura Sinica, 2020, 53(9): 1730-1742.
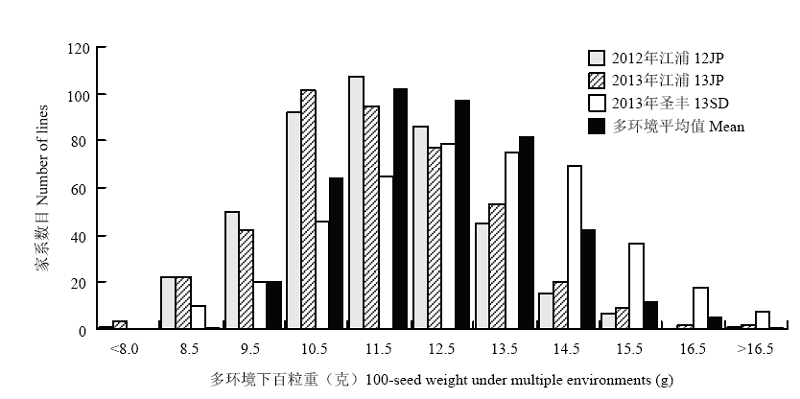
图1
多环境下NJRIKY群体百粒重的频数分布图 Mean表示各家系3个环境百粒重平均数"
表1
多环境下NJRIKY群体百粒重的次数分布和描述统计"
| 环境 Environment | 亲本 Parent (g) | 重组自交系群体 RIL | |||||
|---|---|---|---|---|---|---|---|
| 科丰1号 Kefeng-1 | 南农1138-2 NN1138-2 | 平均值 Mean | 最小值 Min. | 最大值 Max. | 遗传变异 系数GCV(%) | 遗传率 h2(%) | |
| 12JP | 8.5 | 15.7 | 11.4 | 7.5 | 16.0 | 13.5 | 93.3 |
| 13JP | 8.8 | 14.9 | 11.6 | 7.6 | 20.6 | 15.5 | 92.5 |
| 13SF | 9.8 | 23.2 | 13.9 | 9.2 | 19.5 | 15.1 | 93.9 |
| 平均 Mean | 9.0 | 17.9 | 12.3 | 8.6 | 17.1 | 12.4 | 85.4 |
表2
NJRIKY群体多环境联合方差分析结果"
| 变异来源 Source of variation | 自由度 DF | 均方 MS | F值 F value | P值 P value |
|---|---|---|---|---|
| 环境 Environment | 2 | 2308.47 | 101.66 | <0.0001 |
| 重复(环境)Replication(Environment) | 6 | 20.08 | 12.59 | <0.0001 |
| 区组(环境×重复)Block(Environment×Replication) | 180 | 1.34 | 1.78 | <0.0001 |
| 家系 Line | 426 | 23.66 | 6.81 | <0.0001 |
| 家系×环境 Line×Environment | 851 | 3.50 | 4.67 | <0.0001 |
| 误差 Error | 2245 | 0.75 | ||
| 合计 Total | 3710 |
表3
NJRIKY群体中利用不同定位方法检测到的百粒重位点结果概要"
| 定位方法 Method | 检测的QTL Detected QTLs | 表型变异解释率 PVE (%) | 已报道的QTL Reported QTLs |
|---|---|---|---|
| RTM-GWAS | |||
| Mapped QTL | 57(19) | 70.78 | 41(84) |
| LC major QTL | 5(5) | 23.30 | |
| SC major QTL | 52(19) | 47.48 | |
| QTL×Env. | 20(13) | 4.20 | |
| Unmapped QTL | 14.52 | ||
| CIM | |||
| Mapped QTL | 14(8) | 56.47 | 13(16) |
| MLM-GWAS | |||
| Mapped QTL | 6(3) | 18.47 | 6(18) |
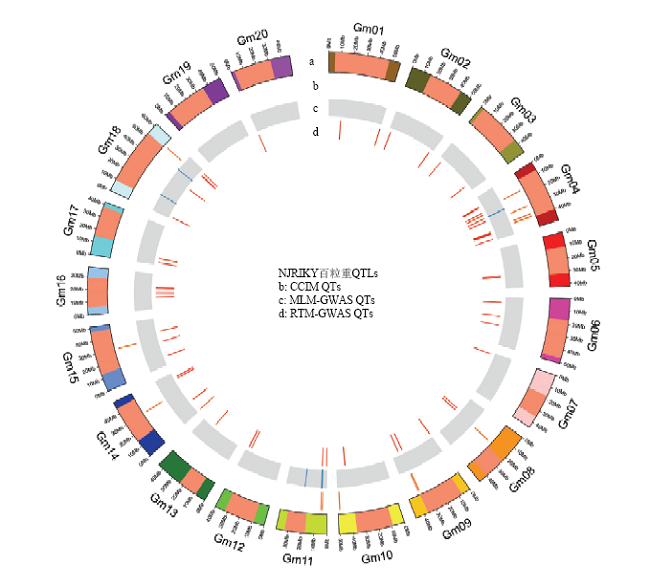
图2
3种定位方法下百粒重的QTL全基因组分布图 a:染色体结构,异染色质区域标为浅红色(单位为Mb)"
表4
NJRIKY群体中在CIM和MLM-GWAS方法下的百粒重QTL"
| 标记Marker | LOD/-lgP | 表型变异解释率PVE (%) | SoyBase QTL |
|---|---|---|---|
| CIM | |||
| Gm04_BLOCK_15191938_15271503 | 3.67 | 2.94 | 5-2 |
| Gm04_BLOCK_36700318_36900222 | 3.08 | 2.55 | 36-15 |
| Gm04_BLOCK_39069193_39185415 | 2.93 | 2.37 | 36-15 |
| Gm04_BLOCK_38868989_39068925 | 2.45 | 2.01 | 36-15 |
| Gm04_BLOCK_29674388_29831299 | 1.96 | 1.59 | 20-2 |
| Gm08_BLOCK_19121233_19314940 | 4.11 | 3.41 | 7-1,34-1,34-13,36-1,37-8 |
| Gm09_BLOCK_35812609_35851884 | 6.81 | 5.76 | 40-4 |
| Gm09_36859571 | 6.97 | 5.86 | 40-4 |
| Gm10_BLOCK_49435374_49524464 | 4.44 | 3.69 | |
| Gm11_3955010 | 7.78 | 6.91 | 37-9 |
| Gm11_BLOCK_5228756_5269867 | 12.39 | 11.03 | 21-1 |
| Gm14_BLOCK_29877119_30044908 | 2.36 | 1.90 | 23-1,13-2 |
| Gm15_BLOCK_29781764_29980832 | 3.69 | 2.95 | 6-1,36-12 |
| Gm18_54211355 | 4.12 | 3.50 | 3-3 |
| 总计Total | 56.47 | ||
| MLM-GWAS | |||
| Gm04_BLOCK_23431433_23626782 | 3.72 | 3.36 | 20-2,5-2,36-15,45-3 |
| Gm11_BLOCK_4611160_4662697 | 3.27 | 2.88 | 37-9 |
| Gm11_BLOCK_5228756_5269867 | 4.03 | 3.69 | 37-9,21-1,22-2,49-2 |
| Gm11_20972740 | 3.12 | 2.73 | 21-1,20-4,20-3,11-1,32-1,4-1,10-3,36-11 |
| Gm18_BLOCK_16884478_16885174 | 3.40 | 3.03 | 2-5 |
| Gm18_45987500 | 3.17 | 2.78 | 27-1 |
| 总计Total | 18.47 |
表5
NJRIKY群体中与百粒重性状关联的QTL位点"
| QTL | 标记 Marker | -lgP | 贡献率 R2 (%) | QTL | 标记 Marker | -lgP | 贡献率 R2 (%) | |
|---|---|---|---|---|---|---|---|---|
| Sw-11-2 | Gm11_BLOCK_5228756_5269867 | 56.22 | 7.57* | Sw-05-1 | Gm05_BLOCK_28628661_28825701 | 7.42 | 0.82* | |
| Sw-09-2 | Gm09_BLOCK_34358756_34538440 | 39.72 | 5.12* | Sw-04-1 | Gm04_BLOCK_10110270_10257050 | 6.78 | 0.74 | |
| Sw-04-7 | Gm04_BLOCK_39928569_39957258 | 29.72 | 3.72* | Sw-06-1 | Gm06_BLOCK_5723355_5918489 | 6.58 | 0.71 | |
| Sw-08-1 | Gm08_16468204 | 28.90 | 3.61 | Sw-06-3 | Gm06_30304358 | 6.52 | 0.71* | |
| Sw-10-1 | Gm10_BLOCK_41285429_41285653 | 26.41 | 3.27 | Sw-15-2 | Gm15_25471398 | 6.31 | 0.68 | |
| Sw-18-5 | Gm18_59805347 | 24.23 | 2.98* | Sw-16-5 | Gm16_BLOCK_23266112_23462965 | 5.98 | 0.64 | |
| Sw-13-1 | Gm13_31358574 | 23.33 | 2.86* | Sw-02-1 | Gm02_BLOCK_2190737_2384686 | 5.72 | 0.61* | |
| Sw-17-1 | Gm17_BLOCK_2938402_3123352 | 20.28 | 2.45 | Sw-04-4 | Gm04_BLOCK_19626939_19826251 | 5.51 | 0.59 | |
| Sw-04-9 | Gm04_BLOCK_40868266_40917625 | 19.75 | 2.38 | Sw-03-2 | Gm03_BLOCK_41020781_41042956 | 5.12 | 0.54 | |
| Sw-16-1 | Gm16_BLOCK_12959169_13153966 | 16.33 | 1.94 | Sw-03-1 | Gm03_3970385 | 4.52 | 0.47 | |
| Sw-01-2 | Gm01_BLOCK_55629182_55799927 | 15.79 | 1.87* | Sw-15-3 | Gm15_36895087 | 4.48 | 0.46 | |
| Sw-18-1 | Gm18_BLOCK_1922631_2035120 | 15.72 | 1.86* | Sw-06-2 | Gm06_20892550 | 4.34 | 0.45* | |
| Sw-14-4 | Gm14_BLOCK_44088379_44288183 | 15.18 | 1.79* | Sw-04-2 | Gm04_BLOCK_10815779_11015107 | 3.97 | 0.40 | |
| Sw-14-1 | Gm14_1033115 | 14.10 | 1.65 | Sw-10-3 | Gm10_BLOCK_41542940_41691982 | 3.68 | 0.37 | |
| Sw-07-1 | Gm07_BLOCK_1944869_1966911 | 12.09 | 1.40 | Sw-01-1 | Gm01_BLOCK_12639088_12838656 | 3.65 | 0.36 | |
| Sw-09-1 | Gm09_BLOCK_264001_463636 | 11.95 | 1.38* | Sw-12-2 | Gm12_BLOCK_37972975_37975786 | 3.46 | 0.34* | |
| Sw-15-1 | Gm15_BLOCK_9079223_9250661 | 11.21 | 1.29* | Sw-14-2 | Gm14_BLOCK_13649138_13846641 | 3.19 | 0.31 | |
| Sw-04-8 | Gm04_BLOCK_40184350_40334324 | 10.84 | 1.24 | Sw-14-3 | Gm14_BLOCK_42062012_42261888 | 2.98 | 0.29 | |
| Sw-18-2 | Gm18_36907653 | 10.77 | 1.23 | Sw-04-11 | Gm04_BLOCK_47626591_47646641 | 2.94 | 0.28 | |
| Sw-10-2 | Gm10_41392627 | 10.09 | 1.15 | Sw-16-2 | Gm16_15575581 | 2.92 | 0.28 | |
| Sw-08-2 | Gm08_BLOCK_19121233_19314940 | 9.71 | 1.10 | Sw-04-10 | Gm04_41805915 | 2.65 | 0.25 | |
| Sw-09-3 | Gm09_BLOCK_39036252_39210642 | 9.27 | 1.04 | Sw-16-4 | Gm16_BLOCK_21928405_22126811 | 2.54 | 0.24 | |
| Sw-11-1 | Gm11_BLOCK_767896_842085 | 8.80 | 0.99 | Sw-04-6 | Gm04_BLOCK_26906733_27091677 | 2.54 | 0.24* | |
| Sw-12-1 | Gm12_BLOCK_35239969_35240158 | 8.77 | 0.98 | Sw-18-4 | Gm18_56862453 | 2.13 | 0.19 | |
| Sw-17-2 | Gm17_BLOCK_7387475_7572446 | 8.11 | 0.90* | Sw-04-3 | Gm04_BLOCK_17249185_17373282 | 2.03 | 0.18 | |
| Sw-16-3 | Gm16_BLOCK_17551378_17602014 | 8.03 | 0.89 | Sw-05-2 | Gm05_30058805 | 1.83 | 0.16* | |
| Sw-18-3 | Gm18_BLOCK_56236139_56414021 | 8.00 | 0.89 | Sw-12-3 | Gm12_BLOCK_38208501_38296133 | 1.61 | 0.13* | |
| Sw-02-2 | Gm02_BLOCK_48637411_48823121 | 7.59 | 0.84 | Sw-20-1 | Gm20_236372 | 1.52 | 0.13 | |
| Sw-04-5 | Gm04_BLOCK_23431433_23626782 | 7.59 | 0.84* | 共计Total | 57 | 70.78 |
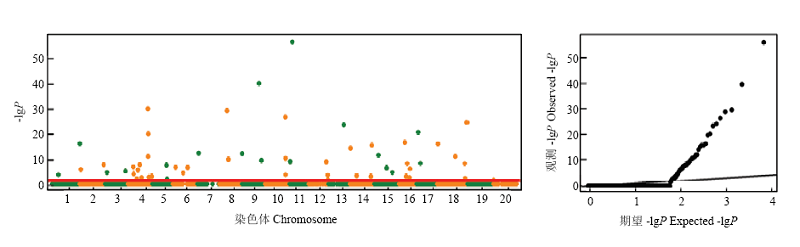
图3
NJRIKY中百粒重关联分析的曼哈顿图和QQ图"
表6
RTM-GWAS方法中检测到的与百粒重相关的候选基因体系"
| QTL | 解释率 R2 (%) | 候选基因 Candidate gene | SNP数目 No. of SNPs | GO注释 GO description |
|---|---|---|---|---|
| Sw-01-2 | 1.87 | Glyma01g45220 | 6 | 胚胎发育Embryo Development |
| Sw-02-1 | 0.61 | Glyma02g02860 | 1 | PSII相关的光收集复合物II过程PSII associated light-harvesting complex II process |
| Sw-02-2 | 0.84 | Glyma02g43960 | 2 | 种子休眠时胚胎发育终止Embryo development ending in seed dormancy |
| Sw-03-1 | 0.47 | Glyma03g04020 | 1 | 多糖分解过程Polysaccharide catabolic process |
| Sw-03-2 | 0.54 | Glyma03g33360 | 1 | 麦芽糖代谢过程Maltose metabolic process |
| Sw-04-1 | 0.74 | Glyma04g11582 | 1 | 转运Transport |
| Sw-04-7 | 3.72 | Glyma04g34070 | 1 | 响应镉离子Response to cadmium- ion |
| Sw-04-9 | 2.38 | Glyma04g34660 | 1 | 脂肪酸代谢过程Fatty acid catabolic process |
| Sw-04-10 | 0.25 | Glyma04g35511 | 1 | 转录的调节,依赖DNA Regulation of transcription, DNA-dependent |
| Sw-04-11 | 0.28 | Glyma04g41810 | 2 | 蛋白质糖基化Protein glycosylation |
| Sw-05-1 | 0.82 | Glyma05g23100 | 1 | |
| Sw-06-1 | 0.71 | Glyma06g07880 | 2 | 种子休眠时胚胎发育终止Embryo development ending in seed dormancy |
| Sw-07-1 | 1.40 | Glyma07g02940 | 3 | ATP分解过程ATP catabolic process |
| Sw-08-1 | 3.61 | Glyma08g21620 | 3 | 分生组织生长调节Regulation of meristem growth |
| Sw-08-2 | 1.10 | Glyma08g24950 | 1 | 细胞生长Cell growth |
| Sw-09-1 | 1.38 | Glyma09g00430 | 4 | 响应脱落酸Response to abscisic acid stimulus |
| Sw-09-3 | 1.04 | Glyma09g32540 | 2 | 毒素分解过程Toxin catabolic process |
| Sw-10-1, | 3.27 | Glyma10g32920 | 2 | 种子发育Seed development |
| Sw-10-2 | 1.15 | 2 | ||
| Sw-10-3 | 0.37 | Glyma10g33160 | 1 | 花器官的形成Floral organ formation |
| Sw-11-1 | 0.99 | Glyma11g01405 | 4 | 鸟苷四磷酸代谢过程Guanosine tetraphosphate metabolic process |
| Sw-11-2 | 7.57 | Glyma11g07430 | 1 | 葡糖醛酸木聚糖代谢过程Glucuronoxylan metabolic process |
| Sw-12-1 | 0.98 | Glyma12g31670 | 2 | 细胞过程规则Abaxial cell fate specification |
| Sw-12-3 | 0.13 | Glyma12g35020 | 4 | 转录正调控Positive regulation of transcription |
| Sw-13-1 | 2.86 | Glyma13g28260 | 1 | |
| Sw-14-1 | 1.65 | Glyma14g01790 | 1 | 多糖生物合成过程Polysaccharide biosynthetic process |
| Sw-14-3 | 0.29 | Glyma14g33820 | 1 | 花发育调节作用Regulation of flower development |
| Sw-14-4 | 1.79 | Glyma14g35260 | 1 | 核转录mRNA分解代谢过程Nuclear-transcribed mRNA catabolic process |
| Sw-15-1 | 1.29 | Glyma15g12410 | 4 | 蛋白质糖基化Protein glycosylation |
| Sw-16-5 | 0.64 | Glyma16g20730 | 1 | 过渡金属离子转运Transition metal ion transport |
| Sw-17-1 | 2.45 | Glyma17g04360 | 1 | |
| Sw-17-2 | 0.90 | Glyma17g09961 | 3 | |
| Sw-18-1 | 1.86 | Glyma18g02940 | 1 | 转录调控Regulation of transcription |
| Sw-18-3 | 0.89 | Glyma18g46517 | 2 | |
| Sw-18-4 | 0.19 | Glyma18g47240 | 1 | 表皮细胞分裂调节Regulation of epidermal cell division |
| Sw-18-5 | 2.98 | Glyma18g50760 | 3 | 多糖生物合成过程Polysaccharide biosynthetic process |
| Sw-20-1 | 0.13 | Glyma20g00565 | 4 | |
| 共计Total | 54.12 | 36 |
| [1] |
SMITH T J, CAMPER H M . Effect of seed size on soybean performance. Agronomy Journal, 1970,67(5):681-684.
doi: 10.1002/(SICI)1097-0215(19960904)67:5&lt;681::AID-IJC15&gt;3.0.CO;2-8 pmid: 8782658 |
| [2] | BURRIS J S, EDJE O T, WAHAB A H . Effects of seed size on seedling performance in soybeans: II. seedling growth and photosynthesis and field performance1. Crop Science, 1973,13(2):207. |
| [3] |
ZENG Z . Precision mapping of quantitative trait loci. Genetics, 1994,136(4):1457-1468.
pmid: 8013918 |
| [4] |
FUJII K, SAYAMA T, TAKAGI K, TAKAGI K, KOSUGE K, OKANO K, KAGA A, ISHINOTO M . Identification and dissection of single seed weight QTLs by analysis of seed yield components in soybean. Breeding Science, 2018,68(2):177-187.
doi: 10.1270/jsbbs.17098 pmid: 29875601 |
| [5] |
ZHANG Y, LI W, LIN Y, ZHANG L, WANG C, XU R . Construction of a high-density genetic map and mapping of QTLs for soybean (Glycine max) agronomic and seed quality traits by specific length amplified fragment sequencing. BMC Genomics, 2018,19(1):641.
doi: 10.1186/s12864-018-5035-9 pmid: 30157757 |
| [6] |
ZHOU Z, JIANG Y, WANG Z, GOU Z, LYU J, LI W, YU Y, SHU L, ZHAO Y, MA Y, FANG C, SHEN Y, LIU T, LI C, LI Q, WU M, WANG M, WU Y, DONG Y, WAN W, WANG X, DING Z, GAO Y, XIANG H, ZHU B, LEE S H, WANG W, TIAN Z . Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nature Biotechnology, 2015,33(4):408-414.
doi: 10.1038/nbt.3096 pmid: 25643055 |
| [7] | ZHANG H, HAO D, SITOE H M, YIN Z, HU Z, ZHANG G, YU D, SINGH R . Genetic dissection of the relationship between plant architecture and yield component traits in soybean (Glycine max) by association analysis across multiple environments. Plant Breeding, 2015,134(5):564-572. |
| [8] |
CONTRERAS-SOTO R I, MORA F, DE OLIVEIRA M A, HIGASHI, W, SCAPIM C A, SCHUSTER I A . Genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PLoS ONE, 2017,12(2):e0171105.
doi: 10.1371/journal.pone.0171105 pmid: 28152092 |
| [9] |
HU Z, ZHANG D, ZHANG G, KAN G, HONG D, YU D . Association mapping of yield-related traits and SSR markers in wild soybean (Glycine soja Sieb. and Zucc.). Breeding Science, 2014,63(5):441-449.
doi: 10.1270/jsbbs.63.441 pmid: 24757383 |
| [10] |
ZHANG J, SONG Q, CREGAN P B, JIANG G . Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean (Glycine max). Theoretical and Applied Genetics, 2016,129(1):117-130.
doi: 10.1007/s00122-015-2614-x pmid: 26518570 |
| [11] |
SONAH H, O'DONOUGHUE L, COBER E, RAJCAN I, BELZILE F . Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnology Journal, 2015,13(2):211-221.
doi: 10.1111/pbi.12249 pmid: 25213593 |
| [12] |
YAN L, HOFMANN N, LI S, FERREIRA M E, SONG B, JIANG G, REN S, QUIGLEY C, FICKUS E, GREGAN P, SONG Q . Identification of QTL with large effect on seed weight in a selective population of soybean with genome-wide association and fixation index analyses. BMC Genomics, 2017,18(1):529.
doi: 10.1186/s12864-017-3922-0 pmid: 28701220 |
| [13] |
WANG J, CHU S, ZHANG H, ZHU Y, CHENG HAO, YU D . Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Scientific Reports, 2016,6:20728.
doi: 10.1038/srep20728 pmid: 26856884 |
| [14] |
COPLEY T R, DUCEPPE M O, O'DONOUGHUE L S . Identification of novel loci associated with maturity and yield traits in early maturity soybean plant introduction lines. BMC Genomics, 2018,19(1):167.
doi: 10.1186/s12864-018-4558-4 pmid: 29490606 |
| [15] |
JING Y, ZHAO X, WANG J, TENG W, QIU L, HAN Y, LI W . Identification of the genomic region underlying seed weight per plant in soybean (Glycine max L. Merr.) via high-throughput single- nucleotide polymorphisms and a genome-wide association study. Frontiers in Plant Science, 2018,9:1392.
doi: 10.3389/fpls.2018.01392 pmid: 30369935 |
| [16] |
HE J, MENG S, ZHAO T, XING G, YANG S, LI Y, GUANG R, LU J, WANG Y, XIA Q, YANG B, GAI J . An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theoretical and Applied Genetics, 2017,130(11):2327-2343.
doi: 10.1007/s00122-017-2962-9 pmid: 28828506 |
| [17] |
LI S, CAO Y, HE J, ZHAO T, GAI J . Detecting the QTL-allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure. Theoretical and Applied Genetics, 2017,130(11):2297-2314.
doi: 10.1007/s00122-017-2960-y pmid: 28799029 |
| [18] |
ZHANG Y, HE J, WANG Y, XING G, ZHAO J, LI Y, YANG S, PALMER R G, ZHAO T, GAI J . Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes. Journal of Experimental Botany, 2015,66(20):6311-6325.
doi: 10.1093/jxb/erv342 pmid: 26163701 |
| [19] |
MENG S, HE J, ZHAO T, XING G, LI Y, YANG S, LU J, WANG Y, GAI J . Detecting the QTL-allele system of seed isoflavone content in Chinese soybean landrace population for optimal cross design and gene system exploration. Theoretical and Applied Genetics, 2016,129(8):1557-1576.
doi: 10.1007/s00122-016-2724-0 pmid: 27189002 |
| [20] |
ZHANG Y, HE J, WANG H, WANG H, MENG S, XING G, LI Y, YANG S, ZHAO J, ZHAO T, GAI J . Detecting the QTL-allele system of seed oil traits using multi-locus genome-wide association analysis for population characterization and optimal cross prediction in soybean. Frontiers in Plant Science, 2018,9:1793.
doi: 10.3389/fpls.2018.01793 pmid: 30568668 |
| [21] | ZHANG Y, HE J, MENG S, LIU M, XING G, LI Y, YANG S, YANG J, ZHAO T, GAI J . Identifying QTL-allele system of seed protein content in Chinese soybean landraces for population differentiation studies and optimal cross predictions. Euphytica, 2018,214(9), 157. |
| [22] |
KHAN M A, TONG F, WANG W, HE J, ZHAO T, GAI J . Analysis of QTL-allele system conferring drought tolerance at seedling stage in a nested association mapping population of soybean [Glycine max (L.) Merr.] using a novel GWAS procedure. Planta, 2018,248(4):947-962.
doi: 10.1007/s00425-018-2952-4 pmid: 29980855 |
| [23] |
MACKAY I, POWELL W . Methods for linkage disequilibrium mapping in crops. Trends in Plant Science, 2007,12(2):57-63.
doi: 10.1016/j.tplants.2006.12.001 pmid: 17224302 |
| [24] |
MALOSETTI M, VAN EEUWIJK F A, BOER M P, CASAS A M, ELIA M, MORALEJO M, BHAT P R, RAMSAY L, MOLINA CONO J L . Gene and QTL detection in a three-way barley cross under selection by a mixed model with kinship information using SNPs. Theoretical and Applied Genetics, 2011,122(8):1605-1616.
doi: 10.1007/s00122-011-1558-z pmid: 21373796 |
| [25] |
PAN L, HE J, ZHAO T, XING G, WANG Y, YU D, CHEN S, GAI J . Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two-stage multi-locus GWAS procedure. Theoretical and Applied Genetics, 2018,131(12):2581-2599.
doi: 10.1007/s00122-018-3174-7 pmid: 30167759 |
| [26] |
RIYAN C, LIM J E, SAMOCHA K E, SOKOLOFF G, ABNEY M, SKOL A D, PALMER A A . Genome-wide association studies and the problem of relatedness among advanced intercross lines and other highly recombinant populations. Genetics, 2010,185(3):1033.
doi: 10.1534/genetics.110.116863 pmid: 20439773 |
| [27] | 王永军, 喻德跃, 章元明, 陈受宜, 盖钧镒 . 重组自交系群体的检测调整方法及其在大豆NJRIKY群体的应用. 作物学报, 2004,30(5):413-418. |
| WANY Y J, YU D Y, ZHANG Y M, CHEN S Y, GAI J Y . Method of evaluation and adjustment of recombinant inbred line population and its application to the soybean RIL population NJRIKY. Acta Agronomica Sinica, 2004,30(5):413-418. (in Chinese) | |
| [28] | 盖钧镒, 邱家驯, 赵团结 . 大豆品种南农493-1和南农1138-2与其衍生新品种的亲缘关系及其育种价值分析. 南京农业大学学报, 1997,20(1):1-8. |
| GAI J Y, QIU J X, ZHAO T J . An analysis of genetic relationship of Nannong493-1 and Nannong 1138-2 with their derivative cultivars and their potential in future breeding. Journal of Nanjing Agricultural University, 1997,20(1):1-8. (in Chinese) | |
| [29] |
MURRAY M G, THOMPSON W F . Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research, 1980,8(19):4321-4325.
doi: 10.1093/nar/8.19.4321 pmid: 7433111 |
| [30] |
ROBERTS A, MCMILLAN L, WANG W, PARKER J, RUSYN I, THREADGILL D . Inferring missing genotypes in large SNP panels using fast nearest-neighbor searches over sliding windows. Bioinformatics, 2007,23(13):401-407.
doi: 10.1093/bioinformatics/btm220 pmid: 17646323 |
| [31] |
YU J M, PRESSOIR G, BRIGGS W H, BI I V, YAMASAKI M, DOEBLEY J F, MCMULLEN M D, GAUT B S, NIELSEN D M, HOLLAND J B, KRESOVICH S, BUCKLER E S . A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics, 2006,38(2):203-208.
doi: 10.1038/ng1702 pmid: 16380716 |
| [32] |
BRADBURY P J, ZHANG Z, KROON D E, CASSTEVENS T M, RAMDOSS V, BUCKER E S . TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics, 2007,23(19):2633-2635.
doi: 10.1093/bioinformatics/btm308 pmid: 17586829 |
| [33] |
STRIMMER K . fdrtool: A versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics, 2008,24(12):1461-1462.
doi: 10.1093/bioinformatics/btn209 pmid: 18441000 |
| [34] | GAI J . Quantitative Inheritance//MALOY S, HUGHES K. eds. Brenner’s Encyclopedia of Genetics. 2nd ed. San Diego: Academic Press, 2013: 18-21. |
| [35] |
LU X, XIONG Q, CHENG T, LI Q, LIU X, BI Y, LI W, ZHANG W, MA B, LAI Y, DU W, MAN W, CHEN S, ZHANG J . A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight. Molecular Plant, 2017,10(5):670-684.
doi: 10.1016/j.molp.2017.03.006 pmid: 28363587 |
| [36] |
GAI J, WANG Y, WU X, CHEN S . A comparative study on segregation analysis and QTL mapping of quantitative traits in plants-with a case in soybean. Frontiers of Agriculture in China, 2007,1(1):1-7.
doi: 10.1007/s11703-007-0001-3 |
| [37] |
ZHANG W K, WANG Y J, LUO G Z, ZHANG J, HE C, WU X, GAI J, CHEN S . QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with EST markers. Theoretical and Applied Genetics, 2004,108(6):1131-1139.
doi: 10.1007/s00122-003-1527-2 pmid: 15067400 |
| [38] |
HAN Y, LI D, ZHU D, LI H, LI X, TENG W, LI W . QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theoretical and Applied Genetics, 2012,125(4):671-683.
doi: 10.1007/s00122-012-1859-x pmid: 22481120 |
| [39] | CHEN Q, ZHANG Z, LIU C, XIN D, QIU H, SHAN D, SHAN C, HU G . QTL analysis of major agronomic traits in soybean. Agricultural Sciences in China, 2007,6(4):399-405. |
| [40] |
WANG P, ZHAO Y, LI Z, HSU C, LIU C, FU L, HOU Y, DU Y, XIE S, ZHANG C, GAO J, CAO M, HUANG X, ZHU Y, TANG K, WANG X, TAO W, XIONG Y, ZHU J . Reciprocal regulation of the TOR kinase and ABA receptor balances plant growth and stress response. Molecular Cell, 2018,69(1):100-112.
doi: 10.1016/j.molcel.2017.12.002 pmid: 29290610 |
| [1] | 彭廷燊, 陆久焱, 吴美林, 严雨欣, 刘宏周, 南文斌, 秦小健, 李明, 龚俊义, 梁永书. 多年生水稻黄糯2号和长白7号产量相关性状的QTL分析[J]. 中国农业科学, 2026, 59(7): 1361-1379. |
| [2] | 李永娟, 张悦彤, 王艺博, 赵长江, 宋洁, 陈雪丽, 姚钦. 生物炭施用对大豆轮连作系统土壤固氮微生物nifH基因丰度及群落组成的影响[J]. 中国农业科学, 2026, 59(6): 1272-1285. |
| [3] | 刘方东, 孙磊, 王吴彬, 赵晋铭, 盖钧镒. 我国大豆种植制度的变更和生态栽培区划调整的建议[J]. 中国农业科学, 2026, 59(3): 486-498. |
| [4] | 蔡廷阳, 朱玉鹏, 李瑞东, 吴宗声, 徐一帆, 宋雯雯, 徐彩龙, 吴存祥. 苗期叶损伤对黄淮海夏大豆光合特性、荚果分布及产量形成的影响[J]. 中国农业科学, 2026, 59(2): 292-304. |
| [5] | 叶美金, 陈家婷, 周界光, 尹丽, 胡欣荣, 兰雨昕, 陈斌, 苏龙兴, 刘家君, 刘天超, 李小雨, 马建. 小麦穗密度主效QTL的鉴定、验证及其遗传效应分析[J]. 中国农业科学, 2026, 59(1): 17-28. |
| [6] | 吴琼, 谢香庭, 王磊, 牟勇, 李进伟. 转基因大豆DBN8205转化体特异性定量PCR方法的研发和验证[J]. 中国农业科学, 2026, 59(1): 29-40. |
| [7] | 陈冰嬬, 唐玉劼, 张丽霞, 周宇飞, 于淼, 石贵山, 王新鼎, 李扬, 高士杰, 陆晓春, 王鼐, 刁现民. 中国粒用杂交高粱的绿色革命[J]. 中国农业科学, 2025, 58(8): 1494-1507. |
| [8] | 严孙辉, 罗程, 陈银基, 庄昕波. 细菌纤维素协同pH偏移处理对大豆分离蛋白凝胶特性与微观结构的影响[J]. 中国农业科学, 2025, 58(6): 1210-1222. |
| [9] | 刘路平, 胡雪洁, 祁金, 陈强, 刘智, 赵田湉, 史晓蕾, 刘兵强, 孟庆民, 张孟臣, 韩天富, 杨春燕. 大豆生育期基因E1和E2的启动子克隆及其表达模式分析[J]. 中国农业科学, 2025, 58(5): 840-850. |
| [10] | 杨永庆, 胡朋举, 宋亚辉, 金欣欣, 苏俏, 王瑾. 超高油花生种质SW9721-3品质性状的QTL定位[J]. 中国农业科学, 2025, 58(4): 635-646. |
| [11] | 郑煜, 陈颐, 遆晋松, 史龙飞, 许校博, 李昱霖, 郭瑞. 烟草不同轮作模式碳足迹及经济效益评价[J]. 中国农业科学, 2025, 58(4): 733-747. |
| [12] | 李璐, 谢庄, 谢可盈, 张瀚, 赵卓文, 向奥妮, 李巧龙, 凌英华, 何光华, 赵芳明. 水稻CSSL-Z492单、双片段代换系构建及粒型QTL的遗传解析[J]. 中国农业科学, 2025, 58(3): 401-415. |
| [13] | 张琦, 薛芙珍, 杨秀洁, 姜苏洋, 黄雪娟, 马佳怡, 张哲文, 徐杰飞. 大豆烟酰胺酶GmNIC1在盐碱胁迫下的功能研究[J]. 中国农业科学, 2025, 58(24): 5128-5142. |
| [14] | 马鹤逍, 葛国龙, 张向前, 路战远, 王满秀, 戎美仁, 师晶晶, 张德健, 孙雪萍. 不同作物轮作系统对土壤易氧化有机碳和碳库活度差异性的影响[J]. 中国农业科学, 2025, 58(24): 5201-5215. |
| [15] | 高春华, 赵海军, 赵逢涛, 孔玮琳, 巨飞燕, 李宗新, 石德杨, 刘苹. 生长调节剂对玉米大豆带状间作下夏玉米茎秆特性与产量的影响[J]. 中国农业科学, 2025, 58(23): 4920-4935. |
|
||