






中国农业科学 ›› 2020, Vol. 53 ›› Issue (9): 1717-1729.doi: 10.3864/j.issn.0578-1752.2020.09.003
所属专题: 专题——限制性两阶段多位点全基因组关联分析法的应用
• 专题:限制性两阶段多位点全基因组关联分析法的应用 • 上一篇 下一篇
郝晓帅1,傅蒙蒙1,刘再东1,贺建波1( ),王燕平2,任海祥2,王德亮3,杨兴勇4,程延喜5,杜维广2,盖钧镒1()
),王燕平2,任海祥2,王德亮3,杨兴勇4,程延喜5,杜维广2,盖钧镒1()
收稿日期:2019-09-09
接受日期:2020-01-02
出版日期:2020-05-01
发布日期:2020-05-13
联系方式:
郝晓帅,E-mail:15850563928@163.com。
基金资助:
XiaoShuai HAO1,MengMeng FU1,ZaiDong LIU1,JianBo HE1(),YanPing WANG2,HaiXiang REN2,DeLiang WANG3,XingYong YANG4,YanXi CHENG5,WeiGuang DU2,JunYi GAI1()
Received:2019-09-09
Accepted:2020-01-02
Published:2020-05-01
Online:2020-05-13
摘要:
【目的】对东北大豆种质群体百粒重性状进行全基因组关联分析,全面解析中国大豆主产区百粒重QTL-等位变异遗传构成,为东北地区大豆籽粒大小遗传改良提供理论基础。【方法】以东北地区育种和生产上常用的290份大豆材料作为试验群体,于2013和2014年在东北第二生态亚区的克山、牡丹江、佳木斯和长春4个地点进行百粒重表型鉴定试验。利用RAD-seq方法对试验群体进行基因组测序分析,对原始SNP数据进行过滤及填补缺失数据后,最终获得了82 966个高质量的SNP标记。根据限制性两阶段多位点全基因组关联分析(restricted two-stage multi-locus genome-wide association analysis,RTM-GWAS)方法,首先构建获得15 546个具有复等位变异的SNPLDB标记,然后使用两阶段多位点模型对百粒重性状进行全基因组关联分析。对检测到的百粒重关联SNPLDB标记位点附近(50 kb范围内)的基因进行分析,根据基因内SNP与SNPLDB标记位点之间关联性的卡方测验,筛选可能与百粒重性状相关的候选基因并进行功能注释。最后基于检测的百粒重QTL-等位变异体系分析了不同熟期组材料间的遗传分化。【结果】试验群体百粒重变异范围为18.3—20.7 g,性状遗传率为92.3%。RTM-GWAS方法共检测到76个与大豆百粒重性状关联的SNPLDB标记位点,其中15个位点主效不显著,另外61个主效显著位点解释了65.40%的表型变异;68个与环境互作效应显著的位点解释了17.46%的表型变异,另外8个位点与环境互作效应不显著。在检测到的76个位点中有34个位点与已报道的30个百粒重QTL重叠,另外42个位点为本研究新检测百粒重位点。基于检测的SNPLDB标记位点,共筛选到137个百粒重相关候选基因,功能注释显示这些候选基因不仅参与大豆百粒重的调节,还参与了初级新陈代谢、蛋白质修饰、物质运输、胁迫响应和信号转导等。对各熟期组间QTL-等位变异的遗传分化分析显示,尽管熟期组间百粒重差异不明显,但其QTL-等位变异遗传结构却发生了新生和汰除的变化。【结论】RTM-GWAS方法能相对全面地解析东北大豆种质群体百粒重QTL-等位变异遗传构成。东北大豆种质群体百粒重由大量QTL调控,且QTL与环境互作效应大,QTL存在丰富的复等位变异。由RTM-GWAS方法建立的QTL-等位变异矩阵为群体遗传及演化研究提供了新工具。
郝晓帅,傅蒙蒙,刘再东,贺建波,王燕平,任海祥,王德亮,杨兴勇,程延喜,杜维广,盖钧镒. 东北大豆种质群体百粒重QTL-等位变异的全基因组解析[J]. 中国农业科学, 2020, 53(9): 1717-1729.
XiaoShuai HAO,MengMeng FU,ZaiDong LIU,JianBo HE,YanPing WANG,HaiXiang REN,DeLiang WANG,XingYong YANG,YanXi CHENG,WeiGuang DU,JunYi GAI. Genome-Wide QTL-Allele Dissection of 100-Seed Weight in the Northeast China Soybean Germplasm Population[J]. Scientia Agricultura Sinica, 2020, 53(9): 1717-1729.
表1
东北大豆种质群体百粒重次数分布及描述统计"
| 类型 Type | 百粒重100-seed weight (g) | N | 平均数 Mean | 变幅 Range | 遗传率 h2 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 9 | 11 | 12 | 14 | 16 | 17 | 19 | 21 | 23 | 24 | 26 | 28 | 29 | 31 | |||||
| 环境Environment | |||||||||||||||||||
| 长春CC | 1 | 1 | 0 | 0 | 9 | 45 | 75 | 85 | 52 | 14 | 4 | 4 | 290 | 20.3 | 9.7-28.2 | 0.642 | |||
| 佳木斯JMS | 1 | 2 | 10 | 3 | 1 | 1 | 17 | 57 | 93 | 68 | 28 | 4 | 4 | 0 | 1 | 290 | 20.7 | 8.1-32.0 | 0.780 |
| 克山KS | 3 | 2 | 0 | 1 | 5 | 45 | 91 | 83 | 49 | 9 | 1 | 0 | 1 | 290 | 18.3 | 6.4-28.2 | 0.777 | ||
| 牡丹江MDJ | 2 | 0 | 0 | 0 | 10 | 48 | 98 | 85 | 39 | 4 | 3 | 0 | 1 | 290 | 19.9 | 6.7-28.9 | 0.726 | ||
| Mean | 1 | 1 | 0 | 0 | 7 | 53 | 89 | 90 | 39 | 5 | 4 | 0 | 1 | 290 | 19.8 | 8.2-29.5 | 0.923 | ||
| 熟期组Maturity | |||||||||||||||||||
| MG0 | 1 | 2 | 25 | 55 | 51 | 16 | 4 | 1 | 155 | 19.9 | 13.6-25.6 | ||||||||
| MG00 | 1 | 0 | 0 | 0 | 6 | 11 | 15 | 10 | 2 | 45 | 20.3 | 9.9-24.8 | |||||||
| MG000 | 3 | 3 | 5 | 3 | 1 | 15 | 20.4 | 18.8-21.8 | |||||||||||
| MGI+II | 2 | 9 | 33 | 25 | 6 | 75 | 19.7 | 15.6-22.7 | |||||||||||
| MG0+00+000 | 1 | 0 | 1 | 2 | 34 | 69 | 71 | 29 | 7 | 1 | 215 | 20.0 | 9.9-25.6 | ||||||
表2
东北大豆种质群体百粒重多年多点联合方差分析"
| 模型Model | 变异来源Source | 自由度DF | 均方MS | F | p |
|---|---|---|---|---|---|
| 基因型×年份×地点 Genotype×Year×Location | 年份Year | 1 | 835.46 | 81.59 | <0.001 |
| 地点Location | 3 | 3156.29 | 493.80 | <0.001 | |
| 区组(年份,地点) Block(Year, Location) | 24 | 2.59 | 1.86 | 0.0068 | |
| 基因型Genotype | 289 | 146.47 | 13.74 | <0.001 | |
| 基因型×年份Genotype×Year | 289 | 9.10 | 2.65 | <0.001 | |
| 基因型×地点Genotype×Location | 867 | 5.22 | 1.41 | <0.001 | |
| 基因型×年份×地点Genotype×Year×Location | 849 | 3.70 | 2.65 | <0.001 | |
| 误差Error | 6791 | 1.40 | |||
| 基因型×环境 Genotype×Environment | 环境Environment | 7 | 1978.57 | 308.84 | <0.001 |
| 区组(环境) Block(Environment) | 24 | 2.59 | 1.86 | 0.0068 | |
| 基因型Genotype | 289 | 147.00 | 28.15 | <0.001 | |
| 基因型×环境Genotype×Environment | 2005 | 5.24 | 3.75 | <0.001 | |
| 误差Error | 6791 | 1.40 |
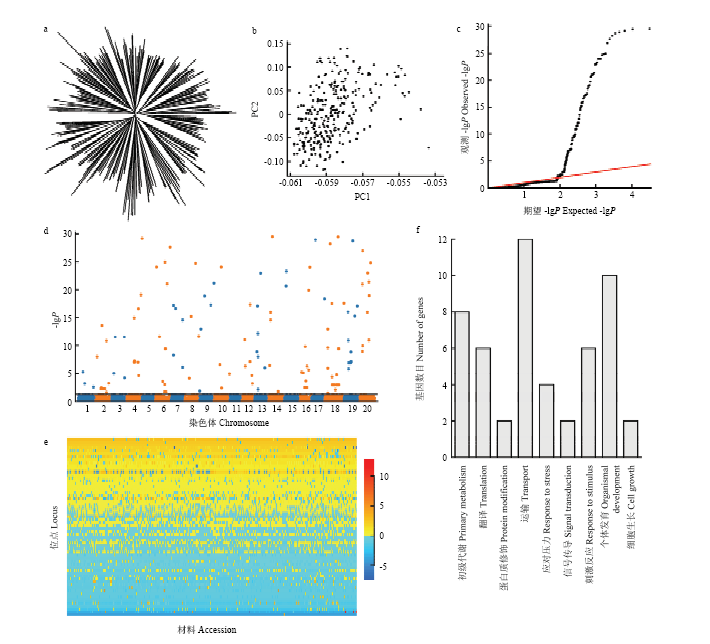
图1
东北大豆种质群体百粒重表型变异的遗传解析 a:Neighbor-joining聚类树;b:遗传相似系数矩阵特征向量散点图,PC1、PC2分别表示前2个特征向量;c:RTM-GWA 方法QQ图。其中-lgP大于30的记为30;d:RTM-GWAS方法Manhattan图;e:东北大豆种质群体百粒重QTL-allele矩阵;f:百粒重候选基因GO生物过程分布"
表3
大豆百粒重显著相关SNPLDB位点"
| QTL | AN | 主效QTL | QTL×Env. a | QTL | AN | 主效QTL | QTL×Env. a | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| -lgP | R2 (%) | -lgP | R2 (%) | -lgP | R2 (%) | -lgP | R2 (%) | |||||
| q-SW-1-1 | 2 | - | - | 6.07 | 0.10 | q-SW-12-1 | 2 | 7.95 | 0.08 | 6.99 | 0.11 | |
| q-SW-1-2 | 2 | - | - | 3.47 | 0.06 | q-SW-12-2 | 8 | 4.34 | 0.07 | 4.63 | 0.24 | |
| q-SW-1-3 | 2 | - | - | 2.55 | 0.05 | q-SW-12-3 | 4 | 5.30 | 0.06 | 20.46 | 0.35 | |
| q-SW-2-1 | 5 | 2.87 | 0.04 | 7.60 | 0.21 | q-SW-12-4 | 2 | 2.27 | 0.02 | 3.83 | 0.07 | |
| q-SW-2-2 | 3 | - | - | 2.85 | 0.06 | q-SW-12-5 | 2 | - | - | 3.50 | 0.06 | |
| q-SW-2-3 | 5 | 203.54 | 2.40 | 11.07 | 0.26 | q-SW-13-1 | 6 | 150.15 | 1.76 | 20.04 | 0.42 | |
| q-SW-2-4 | 2 | - | - | 2.29 | 0.05 | q-SW-13-2 | 7 | 76.04 | 0.90 | 46.13 | 0.80 | |
| q-SW-2-5 | 2 | 13.60 | 0.14 | - | - | q-SW-13-3 | 5 | 28.30 | 0.33 | 14.53 | 0.31 | |
| q-SW-2-6 | 2 | 3.00 | 0.03 | - | - | q-SW-13-4 | 4 | 102.12 | 1.16 | 10.74 | 0.23 | |
| q-SW-3-1 | 3 | 5.73 | 0.06 | - | - | q-SW-14-1 | 7 | - | - | 7.12 | 0.26 | |
| q-SW-3-2 | 6 | 3.19 | 0.05 | 12.21 | 0.31 | q-SW-14-2 | 6 | 192.85 | 2.28 | 19.05 | 0.41 | |
| q-SW-3-3 | 5 | 307.65 | 4.26 | 18.50 | 0.37 | q-SW-14-3 | 2 | 63.12 | 0.69 | - | - | |
| q-SW-3-4 | 4 | - | - | 4.81 | 0.14 | q-SW-14-4 | 2 | - | - | 2.26 | 0.05 | |
| q-SW-4-1 | 6 | 19.08 | 0.24 | 5.81 | 0.21 | q-SW-15-1 | 6 | 34.07 | 0.41 | 7.51 | 0.24 | |
| q-SW-4-2 | 5 | 14.91 | 0.18 | 10.54 | 0.26 | q-SW-15-2 | 6 | 40.33 | 0.48 | 15.05 | 0.35 | |
| q-SW-4-3 | 2 | 307.65 | 10.56 | - | - | q-SW-16-1 | 3 | 213.17 | 2.49 | 3.98 | 0.10 | |
| q-SW-4-4 | 6 | 59.53 | 0.70 | 12.31 | 0.31 | q-SW-16-2 | 4 | - | - | 11.03 | 0.23 | |
| q-SW-6-1 | 2 | 35.63 | 0.38 | 3.29 | 0.06 | q-SW-16-3 | 2 | 2.74 | 0.02 | 2.41 | 0.05 | |
| q-SW-6-2 | 6 | 8.46 | 0.11 | 8.63 | 0.26 | q-SW-16-4 | 5 | 20.46 | 0.24 | 14.44 | 0.31 | |
| q-SW-6-3 | 2 | 2.61 | 0.02 | 2.41 | 0.05 | q-SW-16-5 | 7 | 29.99 | 0.37 | 19.95 | 0.45 | |
| q-SW-6-4 | 7 | 29.03 | 0.36 | 23.57 | 0.50 | q-SW-17-1 | 5 | 53.76 | 0.62 | 7.77 | 0.21 | |
| q-SW-6-5 | 8 | 7.57 | 0.12 | 6.58 | 0.27 | q-SW-17-2 | 5 | 2.98 | 0.04 | 26.98 | 0.48 | |
| q-SW-6-6 | 3 | 35.92 | 0.40 | 3.54 | 0.09 | q-SW-18-1 | 5 | 145.14 | 1.69 | 4.18 | 0.16 | |
| q-SW-6-7 | 4 | 49.75 | 0.56 | 8.91 | 0.20 | q-SW-18-2 | 2 | - | - | 2.29 | 0.05 | |
| q-SW-7-1 | 5 | 12.92 | 0.16 | 15.52 | 0.33 | q-SW-18-3 | 6 | 262.05 | 3.16 | 31.38 | 0.57 | |
| q-SW-7-2 | 2 | 147.28 | 1.66 | 2.36 | 0.05 | q-SW-18-4 | 7 | 17.23 | 0.22 | 13.01 | 0.35 | |
| q-SW-7-3 | 6 | 11.66 | 0.15 | 10.25 | 0.28 | q-SW-18-5 | 2 | - | - | 5.50 | 0.09 | |
| q-SW-8-1 | 2 | - | - | 4.40 | 0.08 | q-SW-18-6 | 4 | 200.62 | 2.35 | 10.23 | 0.22 | |
| q-SW-8-2 | 3 | 90.82 | 1.02 | 4.07 | 0.10 | q-SW-18-7 | 5 | 228.35 | 2.71 | 88.81 | 1.24 | |
| q-SW-8-3 | 2 | - | - | - | - | q-SW-19-1 | 8 | 15.85 | 0.21 | 10.59 | 0.32 | |
| q-SW-8-4 | 2 | 45.35 | 0.49 | - | - | q-SW-19-2 | 7 | 178.86 | 2.12 | 14.09 | 0.37 | |
| q-SW-8-5 | 4 | 114.44 | 1.31 | 8.39 | 0.19 | q-SW-19-3 | 7 | 54.88 | 0.64 | 16.95 | 0.38 | |
| q-SW-9-1 | 2 | - | - | 2.12 | 0.05 | q-SW-19-4 | 6 | 74.96 | 0.87 | 11.80 | 0.30 | |
| q-SW-9-2 | 6 | 8.40 | 0.11 | 10.50 | 0.28 | q-SW-19-5 | 5 | 23.13 | 0.27 | 5.86 | 0.18 | |
| q-SW-9-3 | 2 | 24.99 | 0.26 | 4.68 | 0.08 | q-SW-20-1 | 8 | 307.65 | 5.77 | 55.70 | 0.97 | |
| q-SW-9-4 | 6 | 59.00 | 0.69 | 17.45 | 0.39 | q-SW-20-2 | 5 | 34.46 | 0.40 | 13.88 | 0.30 | |
| q-SW-9-5 | 6 | 95.80 | 1.11 | 14.16 | 0.34 | LC-QTL | 83 | 18 | 52.15 | |||
| q-SW-10-1 | 5 | 6.28 | 0.08 | 3.34 | 0.14 | SC-QTL | 205 | 43 | 13.25 | |||
| q-SW-10-2 | 2 | 307.65 | 4.35 | 7.36 | 0.11 | 总Total | 328 | 61 | 65.40 | 68 | 17.46 | |
| q-SW-10-3 | 2 | 89.96 | 0.99 | - | - | |||||||
表4
百粒重性状相关大效应QTL和候选基因"
| QTL | R2 (%) | 候选基因 Candidate gene | 基因本体生物学过程 Gene ontology biological process |
|---|---|---|---|
| q-SW-3-3 | 4.43 | Glyma03g31790 | 囊泡介导的运输Vesicle-mediated transport |
| Glyma03g31810 | 线粒体mRNA修饰Mitochondrial mRNA modification | ||
| Glyma03g31820 | 微管细胞骨架组织Microtubule cytoskeleton organization | ||
| Glyma03g31940 | 甲壳素响应Response to chitin | ||
| Glyma03g32040 | 高尔基体内囊泡介导转运Intra-Golgi vesicle-mediated transport | ||
| q-SW-4-3 | 10.93 | Glyma04g38830 | 细胞分裂素代谢Cytokinin metabolic |
| Glyma04g38870 | 甲基转移酶活性Methyltransferase activity | ||
| Glyma04g38955 | 糖介导的信号通路Sugar mediated signaling pathway | ||
| q-SW-8-5 | 1.36 | Glyma08g44800 | RRNA加工RRNA processing |
| Glyma08g44820 | 蛋白水解Proteolysis | ||
| Glyma08g44960 | 未知Unknown | ||
| Glyma08g44921 | 跨膜运输Transmembrane transport | ||
| q-SW-9-5 | 1.16 | Glyma09g41070 | 液泡运输Vacuolar transport |
| Glyma09g41140 | 肌醇六磷酸磷酸酯的生物合成过程Myo-inositol hexakisphosphate biosynthetic process | ||
| Glyma09g41150 | 胚胎发育以种子休眠结束Embryo development ending in seed dormancy | ||
| Glyma09g41260 | 氧化应激响应Response to oxidative stress | ||
| Glyma09g41320 | 鸟嘌呤运输Guanine transport | ||
| Glyma09g41121 | 未知Unknown | ||
| q-SW-13-1 | 1.83 | Glyma13g08170 | 翻译调控Regulation of translation |
| q-SW-13-4 | 1.21 | Glyma13g29011 | 种子萌发Seed germination |
| q-SW-14-2 | 2.37 | Glyma14g08040 | 嘧啶核糖核苷酸生物合成Pyrimidine ribonucleotide biosynthetic |
| Glyma14g08050 | 缺氧响应Response to hypoxia | ||
| Glyma14g08070 | 种子萌发正调控Positive regulation of seed germination | ||
| Glyma14g08220 | 脱落酸应激响应Response to abscisic acid stimulus | ||
| Glyma14g08075 | 未知Unknown | ||
| Glyma14g08145 | 未知Unknown | ||
| q-SW-16-1 | 2.58 | Glyma16g06320 | 未知Unknown |
| q-SW-18-1 | 1.75 | Glyma18g10460 | 新陈代谢Metabolic |
| Glyma18g10470 | 防御反应Defense response | ||
| q-SW-18-3 | 3.28 | Glyma18g16720 | 蛋白质折叠Protein folding |
| Glyma18g16761 | 蛋白水解Proteolysis | ||
| q-SW-18-6 | 2.44 | Glyma18g36455 | 未知Unknown |
| q-SW-18-7 | 2.81 | Glyma18g52250 | 盐胁迫响应Response to salt stress |
| Glyma18g52260 | 转录调控Regulation of transcription | ||
| Glyma18g52290 | 碳水化合物代谢Carbohydrate metabolic | ||
| Glyma18g52350 | Basipetal生长素运输Basipetal auxin transport |
表5
百粒重QTL-等位变异在熟期组间的变化"
| QTL | a1 | a2 | a3 | a4 | a5 | a6 | a7 | a8 | QTL | a1 | a2 | a3 | a4 | a5 | a6 | a7 | a8 | QTL | a1 | a2 | a3 | a4 | a5 | a6 | a7 | a8 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1-1 | yz | 7-3 | X | yz | 14-4 | yz | |||||||||||||||||||||||||||||||||
| 1-2 | y | 8-1 | z | 15-1 | X | ||||||||||||||||||||||||||||||||||
| 1-3 | yz | 8-2 | z | 15-2 | z | z | |||||||||||||||||||||||||||||||||
| 2-1 | z | z | 8-3 | XYZ | yz | XZ | 16-1 | ||||||||||||||||||||||||||||||||
| 2-2 | z | yz | 8-4 | xyz | XYZ | XYZ | XYZ | XYZ | xyz | 16-2 | XY | yz | |||||||||||||||||||||||||||
| 2-3 | X | XY | z | 8-5 | xyz | xyz | xyz | xyz | 16-3 | ||||||||||||||||||||||||||||||
| 2-4 | XZ | 9-1 | z | 16-4 | yz | ||||||||||||||||||||||||||||||||||
| 2-5 | z | 9-2 | xy | z | 16-5 | yz | z | ||||||||||||||||||||||||||||||||
| 2-6 | yz | 9-3 | XY | 17-1 | XY | z | |||||||||||||||||||||||||||||||||
| 3-1 | XY | XYZ | Y | 9-4 | XYZ | y | z | 17-2 | z | y | |||||||||||||||||||||||||||||
| 3-2 | X | XZ | 9-5 | yz | 18-1 | XYZ | y | z | |||||||||||||||||||||||||||||||
| 3-3 | yz | yz | 10-1 | X | XY | 18-2 | z | ||||||||||||||||||||||||||||||||
| 3-4 | z | y | 10-2 | X | 18-3 | y | z | z | yz | ||||||||||||||||||||||||||||||
| 4-1 | yz | yz | yz | z | 10-3 | z | 18-4 | yz | yz | yz | XY | ||||||||||||||||||||||||||||
| 4-2 | z | yz | yz | 12-1 | 18-5 | yz | |||||||||||||||||||||||||||||||||
| 4-3 | z | 12-2 | z | XZ | yz | z | z | yz | 18-6 | yz | yz | y | |||||||||||||||||||||||||||
| 4-4 | YZ | yz | xz | z | z | 12-3 | yz | z | 18-7 | z | |||||||||||||||||||||||||||||
| 6-1 | 12-4 | yz | 19-1 | X | |||||||||||||||||||||||||||||||||||
| 6-2 | z | X | 12-5 | z | 19-2 | z | xyz | z | |||||||||||||||||||||||||||||||
| 6-3 | XZ | 13-1 | z | yz | z | 19-3 | XY | XY | |||||||||||||||||||||||||||||||
| 6-4 | z | yz | z | 13-2 | z | yz | XZ | z | 19-4 | yz | yz | z | YZ | ||||||||||||||||||||||||||
| 6-5 | yz | y | yz | y | yz | 13-3 | z | XYZ | z | 19-5 | z | ||||||||||||||||||||||||||||
| 6-6 | yz | yz | 13-4 | z | yz | 20-1 | XZ | X | z | y | yz | XY | X | ||||||||||||||||||||||||||
| 6-7 | xyz | 14-1 | yz | z | z | 20-2 | y | y | |||||||||||||||||||||||||||||||
| 7-1 | XYZ | z | 14-2 | XYZ | XY | yz | |||||||||||||||||||||||||||||||||
| 7-2 | 14-3 | xy | |||||||||||||||||||||||||||||||||||||
| 熟期组 Maturity group | 等位变异总数 Total allele | 继承等位变异 Inherent allele | 变化等位变异 Changed allele | 新生等位变异 Emerged allele | 汰除等位变异 Excluded allele | ||||||||||||||||||||||||||||||||||
| Allele no. | QTL no. | Allele no. | QTL no. | Allele no. | QTL no. | Allele no. | QTL no. | Allele no. | QTL no. | ||||||||||||||||||||||||||||||
| Ⅰ+Ⅱ | 292 (147, 145) | 76 | |||||||||||||||||||||||||||||||||||||
| 0 vs.Ⅰ+Ⅱ | 321 (162, 159) | 76 | 287 (144, 143) | 76 | 39 (21,18) | 30 | 34 (18,16) | 25 | 5 (3,2) | 5 | |||||||||||||||||||||||||||||
| 00 vs. 0 | 250 (125,125) | 76 | 247 (123, 124) | 76 | 77(41,36) | 49 | 3 (2,1) | 2 | 74 (39,35) | 49 | |||||||||||||||||||||||||||||
| 000 vs. 00 | 208 (105,103) | 76 | 189 (96,93) | 76 | 80(38,42) | 52 | 19 (9,10) | 17 | 61(29,32) | 44 | |||||||||||||||||||||||||||||
| 0+00+000 vs.Ⅰ+Ⅱ | 324 (163, 161) | 76 | 288 (144, 144) | 76 | 40 (22,18) | 31 | 36 (19,17) | 27 | 4(3,1) | 4 | |||||||||||||||||||||||||||||
| [1] |
陈强, 闫龙, 冯燕, 邓莹莹, 侯文焕, 刘青, 刘兵强, 杨春燕, 张孟臣 . 大豆百粒重QTL定位及多样性评价. 中国农业科学, 2016,49(9):1646-1656.
doi: 10.3864/j.issn.0578-1752.2016.09.002 |
|
CHEN Q, YAN L, FENG Y, DENG Y Y, HOU W H, LIU Q, LIU B Q, YANG C Y, ZHANG M C . QTL Mapping and diversity evaluation of soybean 100-seed weight. Scientia Agricultura Sinica, 2016,49(9):1646-1656. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2016.09.002 |
|
| [2] |
LU X, XIONG Q, CHENG T, LI Q T, LIU X L, BI Y D, LI W, ZHANG W K, MA B, LAI Y C, DU W G, MAN W Q, CHEN S Y, ZHANG J S . A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight. Molecular Plant, 2017,10(5):670-684.
doi: 10.1016/j.molp.2017.03.006 pmid: 28363587 |
| [3] | 汪霞, 李广军, 李河南, 艮文全, 章元明 . 大豆百粒重QTL定位. 作物学报, 2010,36(10):1674-1682. |
| WANG X, LI G J, LI H N, GEN W Q, ZHANG Y M . QTL mapping for soybean 100-seed weight. The Crop Journal, 2010,36(10):1674-1682. (in Chinese) | |
| [4] | 齐照明, 孙亚男, 陈立君, 郭强, 刘春燕, 胡国华, 陈庆山 . 基于Meta分析的大豆百粒重的QTLs定位. 中国农业科学, 2009,42(11):3795-3803. |
| QI Z M, SUN Y N, CHEN L J, GUO Q, LIU C Y, HU G H, CHEN Q S . Meta-analysis of 100-seed weight QTLs in soybean. Scientia Agricultura Sinica, 2009,42(11):3795-3803. (in Chinese) | |
| [5] |
MIAN M A R, BAILEY M A, TAMULONIS J P, SHIPE E R, CARTER T E, PARROTT W A, ASHLEY D A, HUSSEY R S, BOERMA H R . Molecular markers associated with seed weight in two soybean populations. Theoretical and Applied Genetics, 1996,93:1011-1016.
doi: 10.1007/BF00230118 pmid: 24162474 |
| [6] | 傅蒙蒙, 王燕平, 任海祥, 王德亮, 包荣军, 杨兴勇, 田忠艳, 曹景举, 傅连舜, 程延喜, 苏江顺, 孙宾成, 杜维广, 赵团结, 盖钧镒 . 东北春大豆熟期组的划分与地理分布. 大豆科学, 2016,35(2):181-192. |
| FU M M, WANG Y P, REN H X, WANG D L, BAO R J, YANG X Y, TIAN Z Y, CAO J J, FU L S, CHENG Y X, SU J S, SUN B C, DU W G, ZHAO T J, GAI J Y . A study on criterion, identification and distribution of maturity groups for spring-sowing soybeans in Northeast China. Soybean Science, 2016,35(2):181-192. (in Chinese) | |
| [7] | MANSUR L M, ORF J H, CHASE K, JARVIK T, CREGAN P B, LARK K G . Genetic mapping of agronomic traits using recombi-nant inbred lines of soybean. Crop Science, 1996,36:1327-1336. |
| [8] |
CSANÁDI G, VOLLMANN J, STIFT G, LELLEY T . Seed quality QTLs identified in a molecular map of early maturing soybean. Theoretical and Applied Genetics, 2001,103:912-919.
doi: 10.1007/s001220100621 |
| [9] | 宛煜嵩 . 大豆遗传图谱的构建及若干农艺性状的 QTL 定位分析[D]. 北京: 中国农业科学院, 2002. |
| WAN Y S . Construction of soybean genetie map and QTL analysis of some agronomic traits[D]. Beijing: Chinese Academy of Agrieultural Sciences, 2002. (in Chinese) | |
| [10] | 孙亚男, 仕相林, 蒋洪蔚, 孙殿军, 辛大伟, 刘春燕, 胡国华, 陈庆山 . 大豆百粒重QTL的上位效应和基因型×环境互作效应. 中国油料作物学报, 2012,34(6):598-603. |
| SUN Y N, SHI X L, JIANG H W, SUN D J, XIN D W, LIU C Y, HU G H, CHEN Q S . Epistatic effects and qE interaction effects of QTLs for 100-seed weight in soybean. Chinese Journal of Oil Crop Sciences, 2012,34(6):598-603. (in Chinese) | |
| [11] |
SUN Y N, PANN J B, SHI X L, DU X Y, LIU Q, QI Z M, JIANG H W, XIN D W, LIU C Y, HU G H, CHEN Q S . Multi-environment mapping and meta-analysis of 100-seed weight in soybase. Molecular Biology Report, 2012,39(10):9435-9443.
doi: 10.1007/s11033-012-1808-4 pmid: 22740134 |
| [12] | KASTOORI R R, JEDLICKA J, GRAEF G L, WATERS B M . Identification of new QTLs for seed mineral, cysteine, and methionine concentrations in soybean [Glycine max (L.) Merr.]. Molecular Breeding, 2014,34(2):431-445. |
| [13] |
KATO S, SAYAMA T, FUJII K, YUMOTO S, KONO Y, HWANG T Y, KIKUCHI A, TAKADA Y, TANAKA Y, SHIRAIWA T, ISHIMOTO M . A major and stable QTL associated with seed weight in soybean across multiple environments and genetic backgrounds. Theoretical and Applied Genetics, 2014,127(6):1365-1374.
doi: 10.1007/s00122-014-2304-0 pmid: 24718925 |
| [14] |
HAO D R, CHENG H, YIN Z T, CUI S Y, ZHANG D, WANG H, YU D Y . Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean (Glycine Max) landraces across multiple environments. Theoretical Applied Genetics, 2012,124(3):447-458.
doi: 10.1007/s00122-011-1719-0 pmid: 21997761 |
| [15] |
ZHOU Z, JIANG Y, WANG Z . Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nature Biotechnology, 2015,33:408.
doi: 10.1038/nbt.3096 pmid: 25643055 |
| [16] |
SONAH H, O’DONOUGHUE L, COBER E, RAJCAN I, BELZILE B . BIdentification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soyabean. Plant Biotechnology Journal, 2015,13(2):211-221.
doi: 10.1111/pbi.12249 pmid: 25213593 |
| [17] | GUPTA P K, ROY J K, PRASAD M . Single nucleotide polymorphisms: A new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants. Current Science, 2010,80(4):524-535. |
| [18] |
NACHMAN M W . Single nucleotide polymorphisms and recombination rate in humans. Trends in Genetics, 2001,17(9):481-485.
doi: 10.1016/s0168-9525(01)02409-x pmid: 11525814 |
| [19] |
ZENG Z B . Precision mapping of quantitative trait loci. Genetics, 1994,136(4):1457-1468.
pmid: 8013918 |
| [20] |
AUDIC S, CLAVERIE J M . The significance of digital gene expression profiles. Genome Research, 1997,7(10):986-995.
doi: 10.1101/gr.7.10.986 pmid: 9331369 |
| [21] |
BENJAMINI Y, DANIEL Y . The control of the false discovery rate in multiple testing under dependency. The Annals of Statistics, 2001,4(29):1165-1188.
doi: 10.1186/1471-2105-9-114 pmid: 18298808 |
| [22] |
HE J B, MENG S, ZHAO T J, XING G N, YANG S P, LI Y, GUAN R Z, LU J J, WANG Y F, XIA Q J, YANG B, GAI J Y . An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theoretical and Applied Genetics, 2017,130(11):2327-2343.
doi: 10.1007/s00122-017-2962-9 pmid: 28828506 |
| [23] |
ZHANG Y H, HE J B, WANG Y F, XING G N, ZHAO J M, LI Y, YANG S P, PALMER R G, ZHAO T J, GAI J Y . Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes. Journal of Experimental Botany, 2015,66(20):6311-6325.
doi: 10.1093/jxb/erv342 pmid: 26163701 |
| [24] |
LI S G, CAO Y C, HE J B, ZHAO T J, GAI J Y . Detecting the QTL‑allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure. Theoretical and Applied Genetics, 2017,130(11):2297-2314.
doi: 10.1007/s00122-017-2960-y pmid: 28799029 |
| [25] |
PAN L Y, HE J B, ZHAO T J, XING G N, WANG Y F, YU D Y, CHEN S Y, GAI J Y . The novel restricted two-stage multi-locus GWAS procedure efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two‑stage multi‑locus GWAS procedure. Theoretical and Applied Genetics, 2018,131(12):2581-2599.
doi: 10.1007/s00122-018-3174-7 pmid: 30167759 |
| [26] | 熊冬金 . 中国大豆育成品种(1923-2005)基于系谱和SSR标记的遗传基础研究[D]. 南京: 南京农工业大学, 2009. |
| XIONG D J . Studies on the genetic bases of Chinese soybean cultivars released durling 1923-2005 based on pedigree and SSR marker analysis[D]. Nanjing: Nanjing Agricultural University, 2009. (in Chinese) | |
| [27] |
ANDOLFATTO P, DAVISON D, EREZYILMAZ D, HU T T, MAST J, SUNAYAMA-MORITA T, STERN D L . Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Research, 2011,21(4):610-617.
doi: 10.1101/gr.115402.110 pmid: 21233398 |
| [28] |
LI R, YU C, LI Y, LAM T W, YIU S M, KRISTIANSEN K, WANG J . SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics, 2009,25(15):1966-1967.
doi: 10.1093/bioinformatics/btp336 pmid: 19497933 |
| [29] | JEREMY S, STEVEN B. C, JESSICA S, JIAN X M, THERESE M, WILLIAM N, DAVID L. H, QI J S, JAY J. T, IANLIN C, DONG X, UFFE H, GREGORY D. M, YEISOO Y, TETSUYA S, TAISHI U, MADAN K. B, DEVINDER S, BABU V, ERIKA L, MYRON P, DAVID G, SHU S Q, DAVID G, KERRIE B, MONTONA F, BRIAN A, DU J C, TIAN Z X, ZHU L C, NAVDEEP G, TRUPTI J, MARC L, ANAND S, ZHANG X C, KAZUO S, HENRY T. N, ROD A. W, PERRY C, JAMES S, JANE G, DAN R, GARY S, RANDY C. S, SCOTT A. J . Genome sequence of the palaeopolyploid soybean. Nature, 2010,463(14):178-183. |
| [30] |
YI X, LIANG Y, HUERTA-SANCHEZ E, JIN X, CUO Z X, POOL J E, XU X, JIANG H, VINCKENBOSCH N, KORNELIUSSEN T S, ZHENG H, LIU T, HE W, LI K, LUO R, NIE X, WU H, ZHAO M, CAO H, ZOU J, SHAN Y, LI S, YANG Q, ASAN, NI P, TIAN G, XU J, LIU X, JIANG T, WU R, ZHOU G, TANG M, QIN J, WANG T, FENG S, LI G, HUASANG, LUOSANG J, WANG W, CHEN F, WANG Y, ZHENG X, LI Z, BIANBA Z, YANG G, WANG X, TANG S, GAO G, CHEN Y, LUO Z, GUSANG L, CAO Z, ZHANG Q, OUYANG W, REN X, LIANG H, ZHENG H, HUANG Y, LI J, BOLUND L, KRISTIANSEN K, LI Y, ZHANG Y, ZHANG X, LI R, LI S, YANG H, NIELSEN R, WANG J . Sequencing of 50 human exomes reveals adaptation to high altitude. Science, 2010,329(5987):75-78.
doi: 10.1126/science.1190371 pmid: 20595611 |
| [31] |
SCHEET P, STEPHENS M . A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. American Journal of Human Genetics, 2006,78(4):629-644.
doi: 10.1086/502802 pmid: 16532393 |
| [32] |
KUMAR S, DUDLEY J, NEI M, TAMURA K . MEGA: A biologist- centric software for evolutionary analysis of DNA and protein sequences. Briefings in Bioinformatics, 2008,9:299-306.
doi: 10.1093/bib/bbn017 pmid: 18417537 |
| [33] |
盖钧镒, 汪越胜, 张孟臣, 王继安, 常汝镇 . 中国大豆品种熟期组划分的研究. 作物学报, 2001,27(3):286-292.
doi: 10.32687/0869-866X-2019-27-3-286-289 pmid: 31251864 |
|
GAI J Y, WANG Y S, ZHANG M C, WANG J A, CHANG R Z . Studies on the classification of maturity groups of soybean in China. Acta Agronomica Sinia, 2001,27(3):286-292. (in Chinese)
doi: 10.32687/0869-866X-2019-27-3-286-289 pmid: 31251864 |
|
| [34] |
COPLEY T R, DUCEPPE M O, O’DONOUGHUE L . SIdentification of novel loci associated with maturity and yield traits in early maturity soybean plant introduction lines. BMC Genomics, 2018,19(1):167.
doi: 10.1186/s12864-018-4558-4 pmid: 29490606 |
| [35] |
WANG J, CHU S, ZHANG H, ZHU Y, CHENG H, YU D Y . Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Scientific Reports, 2016,6:20728.
doi: 10.1038/srep20728 pmid: 26856884 |
| [36] |
CONTRERAS-SOTO R I, MORA F, OLIVEIRA M A R, HIGASHI W, SCAPIM C A, SCHUSTER I . A genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PLoS ONE, 2017,12(2):e0171105.
doi: 10.1371/journal.pone.0171105 pmid: 28152092 |
| [37] |
ZHANG J, SONG Q, CREGAN P B, JIANG J L . Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean (Glycine max) Theoretical and Applied Genetics, 2016,129:117.
doi: 10.1007/s00122-015-2614-x pmid: 26518570 |
| [1] | 叶美金, 吴雷, Lohani Md Nahibuzzaman, 尹丽, 胡欣荣, 刘亚西, 蒋云峰, 陈国跃, 蒲至恩, 李阳, 李婷, 邹亚亚, 吴佳怡, 马建. 基于GWAS的中国地方小麦成熟胚大小位点的鉴定及其遗传效应解析[J]. 中国农业科学, 2026, 59(6): 1157-1171. |
| [2] | 李永娟, 张悦彤, 王艺博, 赵长江, 宋洁, 陈雪丽, 姚钦. 生物炭施用对大豆轮连作系统土壤固氮微生物nifH基因丰度及群落组成的影响[J]. 中国农业科学, 2026, 59(6): 1272-1285. |
| [3] | 刘方东, 孙磊, 王吴彬, 赵晋铭, 盖钧镒. 我国大豆种植制度的变更和生态栽培区划调整的建议[J]. 中国农业科学, 2026, 59(3): 486-498. |
| [4] | 王勇胜, 牛丽, 王长杰, 马立花, 廉潇潇, 孟亚雄, 马小乐, 姚立蓉, 张宏, 杨轲, 李葆春, 王化俊, 司二静, 汪军成. 冬小麦千粒重的全基因组关联分析及候选基因预测[J]. 中国农业科学, 2026, 59(3): 499-514. |
| [5] | 蔡廷阳, 朱玉鹏, 李瑞东, 吴宗声, 徐一帆, 宋雯雯, 徐彩龙, 吴存祥. 苗期叶损伤对黄淮海夏大豆光合特性、荚果分布及产量形成的影响[J]. 中国农业科学, 2026, 59(2): 292-304. |
| [6] | 吴琼, 谢香庭, 王磊, 牟勇, 李进伟. 转基因大豆DBN8205转化体特异性定量PCR方法的研发和验证[J]. 中国农业科学, 2026, 59(1): 29-40. |
| [7] | 李云丽, 刁邓超, 刘雅睿, 孙玉晨, 孟祥宇, 邬陈芳, 汪妤, 吴建辉, 李春莲, 曾庆东, 韩德俊, 郑炜君. 小麦苗期耐热性全基因组关联分析[J]. 中国农业科学, 2025, 58(9): 1663-1683. |
| [8] | 严孙辉, 罗程, 陈银基, 庄昕波. 细菌纤维素协同pH偏移处理对大豆分离蛋白凝胶特性与微观结构的影响[J]. 中国农业科学, 2025, 58(6): 1210-1222. |
| [9] | 高岩浩, 王婷婷, 白卫卫, 杜兴杰, 刘贤, 秦本源, 付彤, 孙宇, 高腾云, 张天留. 脂质组与转录组联合揭示南阳牛不同肌肉组织脂质特征的差异表达模式[J]. 中国农业科学, 2025, 58(6): 1239-1258. |
| [10] | 刘路平, 胡雪洁, 祁金, 陈强, 刘智, 赵田湉, 史晓蕾, 刘兵强, 孟庆民, 张孟臣, 韩天富, 杨春燕. 大豆生育期基因E1和E2的启动子克隆及其表达模式分析[J]. 中国农业科学, 2025, 58(5): 840-850. |
| [11] | 郑煜, 陈颐, 遆晋松, 史龙飞, 许校博, 李昱霖, 郭瑞. 烟草不同轮作模式碳足迹及经济效益评价[J]. 中国农业科学, 2025, 58(4): 733-747. |
| [12] | 周广飞, 马亮, 马璐, 张舒钰, 章慧敏, 宋旭东, 张振良, 陆虎华, 郝德荣, 冒宇翔, 薛林, 陈国清. 玉米苞叶性状全基因组关联分析[J]. 中国农业科学, 2025, 58(3): 431-442. |
| [13] | 张琦, 薛芙珍, 杨秀洁, 姜苏洋, 黄雪娟, 马佳怡, 张哲文, 徐杰飞. 大豆烟酰胺酶GmNIC1在盐碱胁迫下的功能研究[J]. 中国农业科学, 2025, 58(24): 5128-5142. |
| [14] | 马鹤逍, 葛国龙, 张向前, 路战远, 王满秀, 戎美仁, 师晶晶, 张德健, 孙雪萍. 不同作物轮作系统对土壤易氧化有机碳和碳库活度差异性的影响[J]. 中国农业科学, 2025, 58(24): 5201-5215. |
| [15] | 高春华, 赵海军, 赵逢涛, 孔玮琳, 巨飞燕, 李宗新, 石德杨, 刘苹. 生长调节剂对玉米大豆带状间作下夏玉米茎秆特性与产量的影响[J]. 中国农业科学, 2025, 58(23): 4920-4935. |
|
||