






中国农业科学 ›› 2022, Vol. 55 ›› Issue (2): 248-264.doi: 10.3864/j.issn.0578-1752.2022.02.002
谢晓宇1( ),王凯鸿1,秦晓晓1,王彩香1(),史春辉1,宁新柱2,杨永林3,秦江鸿3,李朝周1,马麒2(),宿俊吉1()
),王凯鸿1,秦晓晓1,王彩香1(),史春辉1,宁新柱2,杨永林3,秦江鸿3,李朝周1,马麒2(),宿俊吉1()
收稿日期:2021-08-13
接受日期:2021-10-26
出版日期:2022-01-16
发布日期:2022-01-26
通讯作者:
王彩香,马麒,宿俊吉
作者简介:谢晓宇,E-mail: 基金资助:
XIE XiaoYu1(),WANG KaiHong1,QIN XiaoXiao1,WANG CaiXiang1(),SHI ChunHui1,NING XinZhu2,YANG YongLin3,QIN JiangHong3,LI ChaoZhou1,MA Qi2(),SU JunJi1()
Received:2021-08-13
Accepted:2021-10-26
Online:2022-01-16
Published:2022-01-26
Contact:
CaiXiang WANG,Qi MA,JunJi SU
摘要:
【目的】吐絮率是反映陆地棉(Gossypium hirsutum L.)早熟性状的重要指标之一,利用全基因组关联分析(genome-wide association study,GWAS)解析吐絮率的QTL(quantitative trait locus)及其遗传效应,为陆地棉早熟性状的分子育种提供理论基础。【方法】利用315份不同陆地棉品种(系)构成的自然群体,在3个环境下对吐絮率进行表型鉴定。同时利用前期构建的9 244个具有复等位变异SNP连锁不平衡区段(SNP linkage disequilibrium block,SNPLDB)分子标记,采用限制性两阶段多位点全基因组关联分析(restricted two-stage multi-locus GWAS,RTM-GWAS)方法,检测与吐絮率显著关联的SNPLDB位点、估算其表型效应值,并建立显著关联位点在群体中QTL-allele矩阵,鉴定稳定关联的主效SNPLDB位点及其优异单倍型。根据2组转录组数据的基因表达量,在显著SNPLDB位点侧翼序列1 Mb基因组范围内挖掘可能与目标性状有关的候选基因。【结果】陆地棉自然群体在3个环境下的吐絮率变异范围为37.78%—100.00%,广义遗传力为67.03%。多环境方差分析表明,吐絮率在基因型、环境及基因型×环境互作间均呈现极显著差异(P<0.001)。通过RTM-GWAS共检测到52个与吐絮率显著关联的SNPLDB位点,共包含179个等位基因/单倍型。其中90个增效等位基因/单倍型的效应值分布范围为0.014—19.43,89个减效等位基因/单倍型的效应值分布范围为-21.49—-0.039。在上述显著关联SNPLDB位点中,6个位点在多环境联合分析和单环境分析中均被检测到,被认为可能是与吐絮率显著关联的稳定SNPLDB位点。通过对上述6个稳定SNPLDB位点不同等位变异对应表型性状的差异显著性分析,鉴定出4个优异等位变异类型,分别为LDB_16_37952328(TT)、LDB_5_96395565(AA)、LDB_16_49503485(TT)和LDB_4_81118668(TT)。进一步分析发现,其优异等位变异的频率分布在中国4个不同生态区的品种(系)间存在差异。此外,在4个稳定主效SNPLDB位点的邻近区域共注释了178个基因,并利用转录组数据分析,预测发现其中23个基因可能是调控陆地棉吐絮率的候选基因。【结论】共鉴定到52个与吐絮率显著相关的SNPLDB位点,其中,有4个SNPLDB为稳定关联的主效位点;预测发现23个基因可能与陆地棉吐絮率有关。
谢晓宇, 王凯鸿, 秦晓晓, 王彩香, 史春辉, 宁新柱, 杨永林, 秦江鸿, 李朝周, 马麒, 宿俊吉. 陆地棉吐絮率的限制性两阶段多位点全基因组关联分析及候选基因预测[J]. 中国农业科学, 2022, 55(2): 248-264.
XIE XiaoYu, WANG KaiHong, QIN XiaoXiao, WANG CaiXiang, SHI ChunHui, NING XinZhu, YANG YongLin, QIN JiangHong, LI ChaoZhou, MA Qi, SU JunJi. Restricted Two-Stage Multi-Locus Genome-Wide Association Analysis and Candidate Gene Prediction of Boll Opening Rate in Upland Cotton[J]. Scientia Agricultura Sinica, 2022, 55(2): 248-264.
表1
陆地棉吐絮率的次数分布和描述统计"
| 环境 Env. | 吐絮率BOR(%) | 平均数 Mean (%) | 变幅 Range (%) | 变异系数 CV (%) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13 | 19 | 25 | 31 | 37 | 43 | 49 | 55 | 61 | 67 | 73 | 79 | 85 | 91 | 97 | N | ||||
| AY-14 | 3 | 5 | 6 | 19 | 31 | 85 | 133 | 33 | 315 | 80.62 | 46.73—90.00 | 9.86 | |||||||
| AY-15 | 4 | 5 | 11 | 23 | 25 | 32 | 35 | 43 | 36 | 22 | 24 | 20 | 19 | 9 | 7 | 315 | 55.99 | 10.42—100.00 | 34.60 |
| SHZ-14 | 1 | 1 | 3 | 16 | 28 | 41 | 63 | 62 | 57 | 34 | 9 | 315 | 70.80 | 37.78—98.05 | 14.42 | ||||
| 综合 Syn. | 1 | 2 | 9 | 24 | 48 | 58 | 80 | 44 | 37 | 12 | 315 | 70.76 | 39.27—91.32 | 14.25 | |||||
表2
陆地棉吐絮率的方差分析"
| 性状 Trait | 方差来源 Variance | 平方和 SS | 均方 MS | F值 F- value | P值 P-value | 遗传力 H2(%) |
|---|---|---|---|---|---|---|
| 吐絮率 BOR | 环境Environment | 321981.19 | 160990.59 | 1084.69 | <0.0001 | 67.03 |
| 基因型Genotype | 288095.59 | 917.50 | 6.18 | <0.0001 | ||
| 环境×基因型Environment ×Genotype | 237515.09 | 378.21 | 2.55 | <0.0001 |
表3
与陆地棉吐絮率显著关联的SNPLDB位点"
| 位点 Loci | 染色体 Chr. | 位置 Position (bp) | -lg(P) | 表型变异 PV (%) | 共同环境a Common environments |
|---|---|---|---|---|---|
| LDB_16_37952328 | 16 (D03) | 37952328 | 46.14 | 1.90 | AY-15 (11.49)、SHZ-14 (5.91) |
| LDB_5_96395565 | 5 (A05) | 96395565 | 22.69 | 0.40 | AY-15 (7.42) |
| LDB_16_49503485 | 16 (D03) | 49503485 | 17.79 | 0.91 | AY-15 (8.49) |
| LDB_10_6908012 | 10 (A10) | 6908012 | 16.70 | 0.87 | AY-15 (8.75) |
| LDB_15_9697358 | 15 (D02) | 9697358 | 13.52 | 1.89 | AY-15 (8.41) |
| LDB_21_22036171 | 21 (D08) | 22036171 | 13.44 | 1.34 | |
| LDB_4_81118668 | 4 (A04) | 81118668 | 11.80 | 0.91 | AY-15 (4.58) |
| LDB_3_6725154_6746183 | 3 (A03) | 6725154 | 11.63 | 1.21 | |
| LDB_1_57527637_57527855 | 1 (A01) | 57527637 | 10.44 | 1.11 | |
| LDB_16_36553916_36554161 | 16 (D03) | 36553916 | 9.81 | 1.72 | |
| LDB_11_119047116 | 11 (A11) | 119047116 | 9.76 | 0.86 | |
| LDB_19_52309050_52309284 | 19 (D06) | 52309050 | 7.97 | 1.54 | |
| LDB_17_51465593 | 17 (D04) | 51465593 | 7.93 | 0.51 | |
| LDB_13_54999733 | 13 (A13) | 54999733 | 7.36 | 0.61 | |
| 位点 Loci | 染色体 Chr. | 位置 Position (bp) | -lg(P) | 表型变异 PV (%) | 共同环境a Common environments |
| LDB_20_6608424 | 20 (D07) | 6608424 | 6.63 | ||
| LDB_7_94884564_94884800 | 7 (A07) | 94884564 | 6.34 | ||
| LDB_19_53788858 | 19 (D06) | 53788858 | 6.25 | 0.27 | |
| LDB_9_58666084_58666088 | 9 (A09) | 58666084 | 6.06 | 1.12 | |
| LDB_6_41226103 | 6 (A06) | 41226103 | 5.52 | 9.00 | |
| LDB_15_46384945_46385146 | 15 (D02) | 46384945 | 5.17 | 0.36 | |
| LDB_15_67139952_67140180 | 15 (D02) | 67139952 | 5.14 | 1.68 | |
| LDB_25_27281705 | 25 (D12) | 27281705 | 5.10 | ||
| LDB_23_6012092 | 23 (D10) | 6012092 | 4.94 | 0.18 | |
| LDB_1_33221331 | 1 (A01) | 33221331 | 4.06 | 0.38 | |
| LDB_12_96771402 | 12 (A12) | 96771402 | 3.84 | 0.97 | |
| LDB_15_65780303 | 15 (D02) | 65780303 | 3.65 | 0.67 | |
| LDB_22_48423583 | 22 (D09) | 48423583 | 3.64 | 0.88 | |
| LDB_14_45778715_45778973 | 14 (D01) | 45778715 | 3.58 | 1.08 | |
| LDB_21_5030313_5030325 | 21 (D08) | 5030313 | 3.55 | 0.15 | |
| LDB_25_39776065 | 25 (D12) | 39776065 | 3.47 | 0.37 | |
| LDB_26_45642548 | 26 (D13) | 45642548 | 3.42 | SHZ-14 (1.77) | |
| LDB_5_30493466 | 5 (A05) | 30493466 | 3.20 | ||
| LDB_12_103902963 | 12 (A12) | 103902963 | 3.15 | 0.39 | |
| LDB_3_42046528_42046540 | 3 (A03) | 42046528 | 2.86 | ||
| LDB_10_70110862_70110882 | 10 (A10) | 70110862 | 2.51 | ||
| LDB_13_72681594_72681843 | 13 (A13) | 72681594 | 2.46 | 0.21 | |
| LDB_19_39327466 | 19 (D06) | 39327466 | 2.43 | ||
| LDB_11_18062977 | 11 (A11) | 18062977 | 2.42 | 0.44 | |
| LDB_8_113962673 | 8 (A08) | 113962673 | 2.30 | ||
| LDB_23_24053424 | 23 (D10) | 24053424 | 2.25 | 0.43 | |
| LDB_21_25090078 | 21 (D08) | 25090078 | 2.19 | 0.47 | |
| LDB_6_108270563 | 6 (A06) | 108270563 | 2.05 | ||
| LDB_25_1835041 | 25 (D12) | 1835041 | 1.86 | 0.18 | |
| LDB_2_4064617 | 2 (A02) | 4064617 | 1.76 | 0.29 | |
| LDB_18_52473670 | 18 (D05) | 52473670 | 1.71 | 0.32 | |
| LDB_13_62696637 | 13 (A13) | 62696637 | 1.62 | 0.30 | |
| LDB_8_8677270 | 8 (A08) | 8677270 | 1.56 | ||
| LDB_24_59986795 | 24 (D11) | 59986795 | 1.48 | ||
| LDB_17_1478345 | 17 (D04) | 1478345 | 1.39 | 0.42 | |
| LDB_5_23314512 | 5 (A05) | 23314512 | 1.34 | ||
| LDB_15_17739938 | 15 (D02) | 17739938 | 1.34 | ||
| LDB_17_53762505_53762526 | 17 (D04) | 53762505 | 1.33 | AY-14 (4.68) |
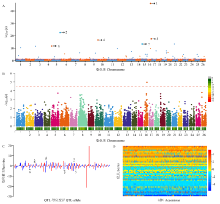
图1
利用RTM-GWAS和MLM-GWAS方法对陆地棉吐絮率的遗传解析 A:RTM-GWAS分析的曼哈顿图;B:MLM-GWAS分析的曼哈顿图;C:所试群体中QTL-allele效应分布;D:所试群体吐絮率的QTL-allele矩阵。A和C图中数字1—6代表筛选的6个稳定关联位点,D中暖色系表示正效应等位基因,冷色系表示负效应等位基因,颜色深度表示效应值的大小"
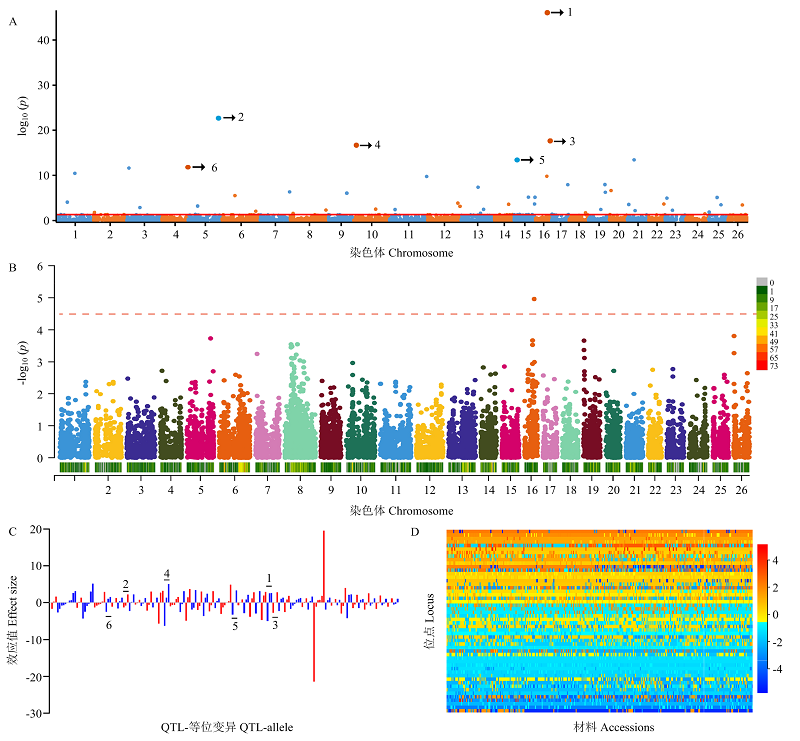

图2
6个稳定显著关联SNPLDB位点不同等位变异的吐絮率比较 BOR:吐絮率。小写字母表示经LSD法多重比较差异显著(P<0.05)。下同"
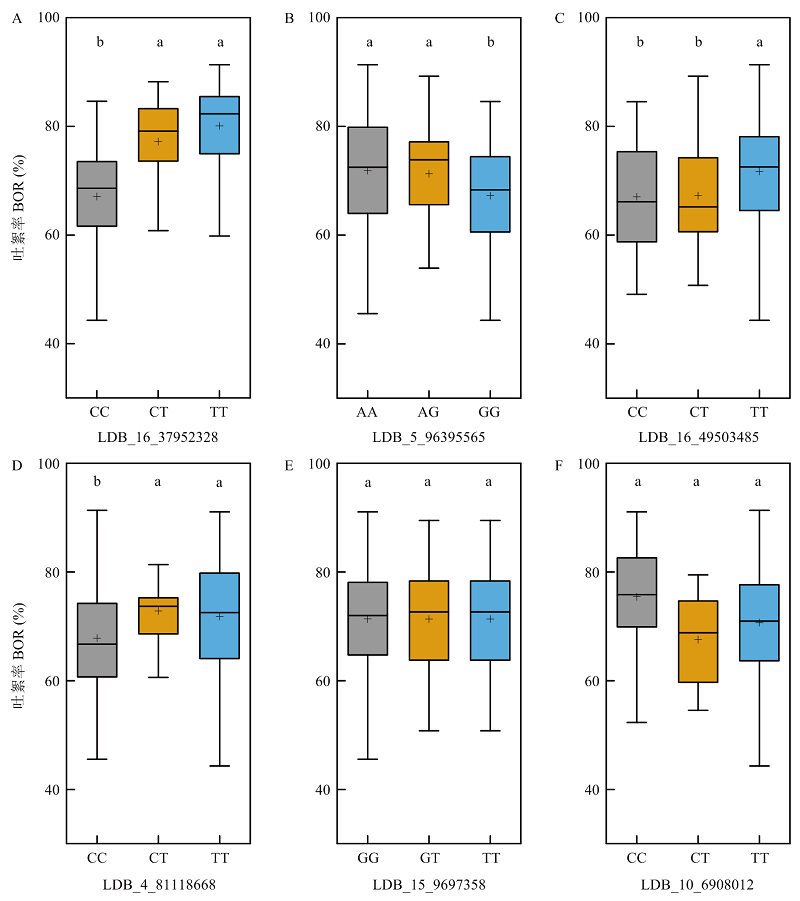
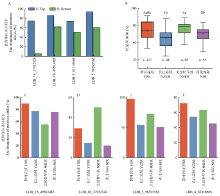
图3
4个稳定关联位点优异等位变异的频率分布 A:4个稳定的SNPLDB位点在高低吐絮率极端材料中优异等位变异的频率比较;B:4个不同地域品种系的吐絮率比较。大写字母表示经LSD法多重比较差异极显著(P<0.01);C—F:4个稳定的SNPLDB位点在4个不同地域品种系间的优异等位变异频率比较。YRR:黄河流域;YZRR:长江流域;NSER:北部特早熟;NIR:西北内陆"
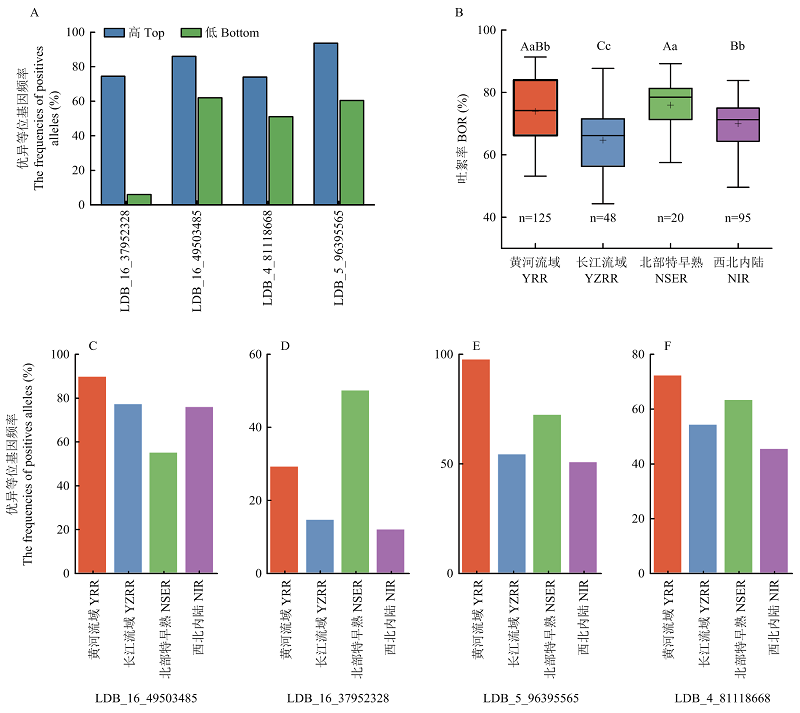

图4
与陆地棉吐絮率相关候选基因的预测 A和B:NAU(A)和CRI(B)的陆地棉RNA-Seq数据不同组织中基因表达模式;C:2个RNA序列数据(NAU和CRI)之间共同基因的韦恩图;D:23个与陆地棉吐絮率相关候选基因表达模式热图。红色代表该基因在某部位或者某时期表达量高,绿色和黑色则表示该基因在某部位或者某时期表达量低"
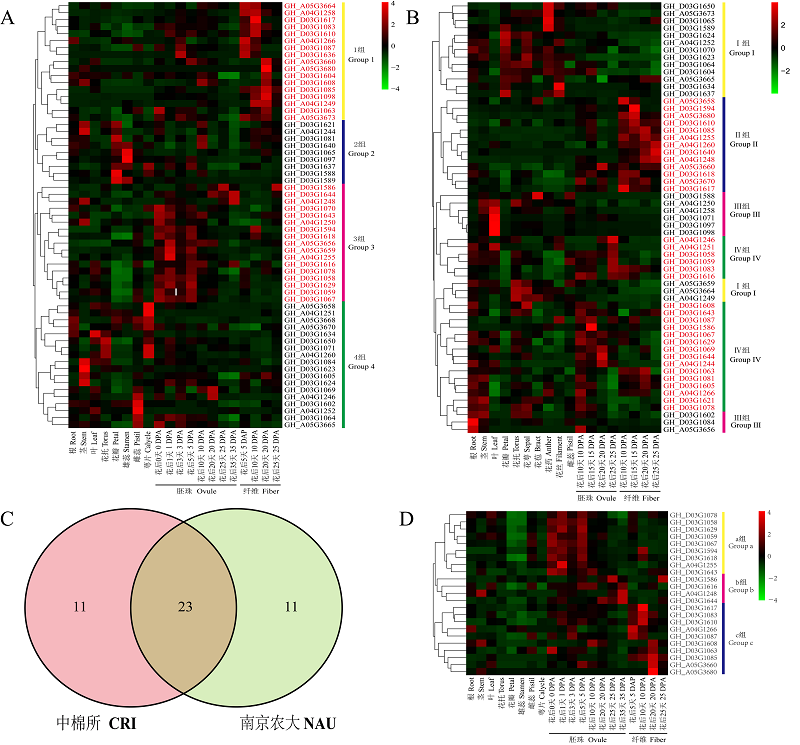
表4
与陆地棉吐絮率相关的候选基因"
| 组别 Group | 候选基因 Candidate gene | 基因名称 Gene name | 基因功能注释 Gene function annotation |
|---|---|---|---|
| a类 Group a | GH_D03G1078 | SWEET10 | 双向糖转运体SWEET10 Bidirectional sugar transporter SWEET10 |
| GH_D03G1058 | slc25a24 | 钙结合线粒体载体蛋白 SCAMC-1 Calcium-binding mitochondrial carrier protein SCaMC-1 | |
| GH_D03G1629 | IRL5 | 植物细胞内Ras-group相关LRR蛋白5 Plant intracellular Ras-group-related LRR protein 5 | |
| GH_D03G1059 | NA | NA | |
| GH_D03G1067 | Bicc1 | 蛋白双尾C同源物1 Protein bicaudal C homolog 1 | |
| GH_D03G1594 | RABD2C | Ras-related蛋白质RABD2c Ras-related protein RABD2c | |
| GH_D03G1618 | NA | 假定的转化酶抑制剂 Putative invertase inhibitor | |
| GH_A04G1255 | RRP6L3 | 蛋白质RRP6-like 3 Protein RRP6-like 3 | |
| GH_D03G1643 | ABCB2 | ABC转运体B家族成员2 ABC transporter B family member 2 | |
| b类 Group b | GH_D03G1586 | HOS1 | E3泛素蛋白连接酶HOS1 E3 ubiquitin-protein ligase HOS1 |
| GH_D03G1616 | BRG3 | 可能与BOI相关的E3泛素蛋白连接酶3 Probable BOI-related E3 ubiquitin-protein ligase 3 | |
| GH_A04G1248 | P3H1 | 脯氨酰3-羟化酶 1 Prolyl 3-hydroxylase 1 | |
| GH_D03G1644 | HSF8 | 热休克因子蛋白HSF8 Heat shock factor protein HSF8 | |
| c类 Group c | GH_D03G1617 | UPF1 | 无意义转录本1同源物的调节器 Regulator of nonsense transcripts 1 homolog |
| GH_D03G1083 | NA | NA | |
| GH_D03G1610 | caskin2 | Caskin-2 | |
| GH_A04G1266 | SPL6 | Squamosa启动子结合样蛋白6 Squamosa promoter-binding-like protein 6 | |
| GH_D03G1087 | PAP27 | 可能失活的紫色酸性磷酸酶27 Probable inactive purple acid phosphatase 27 | |
| GH_D03G1608 | At2g01680 | 含锚蛋白重复蛋白At2g01680 Ankyrin repeat-containing protein At2g01680 | |
| GH_D03G1063 | GLO1 | 过氧化物酶体(S)-2-羟基氧化酶 Peroxisomal (S)-2-hydroxy-acid oxidase GLO1 | |
| GH_D03G1085 | IRX15 | 蛋白质不规则木质部15 Protein IRREGULAR XYLEM 15 | |
| GH_A05G3660 | At3g52300 | ATP合酶亚基d,线粒体 ATP synthase subunit d, mitochondrial | |
| GH_A05G3680 | NA | NA |
| [1] | 喻树迅, 黄祯茂. 短季棉品种早熟性构成因素的遗传分析. 中国农业科学, 1990, 23(6):48-54. |
| YU S X, HUANG Z M. Genetic analysis of precocious factors of cotton varieties in the short season. Scientia Agricultura Sinica, 1990, 23(6):48-54. (in Chinese) | |
| [2] | 宋美珍, 喻树迅, 范术丽, 原日红, 黄祯茂. 短季棉主要农艺性状的遗传分析. 棉花学报, 2005, 17(2) : 94-98. |
| SONG M Z, YU S X, FAN S L, YUAN R H, HUANG Z M. Genetic analysis of main agronomic traits in short season upland cotton. Cotton Science, 2005, 17(2):94-98. (in Chinese) | |
| [3] | 李轲, 李志博, 魏亦农. 棉花早熟性状的相关性分析和QTL定位. 新疆农业科学, 2010, 47(1):78-81. |
| LI K, LI Z B, WEI Y N. Correlation of early maturing traits and QTL mapping in cotton. Xinjiang Agricultural Sciences, 2010, 47(1):78-81. (in Chinese) | |
| [4] | 高玉虹, 姜艳丽, 李朋波, 宋建中, 皇甫张龙, 胡晓丽, 黄晋玲, 石跃进. 棉花早熟性QTL定位研究进展. 山西农业科学, 2012, 40(1):84-86. |
| GAO Y H, JIANG Y L, LI P B, SONG J Z, HUANGFU Z L, HU X L, HUANG J L, SHI Y J. Research progress in QTL mapping of early maturing traits in cotton. Journal of Shanxi Agricultural Sciences, 2012, 40(1):84-86. (in Chinese) | |
| [5] | 赵树琪, 庞朝友, 魏恒玲, 王寒涛, 李黎贝, 宿俊吉, 范术丽, 喻树迅. 陆地棉早熟性状多世代联合遗传分析. 棉花学报, 2017, 29(2):119-127. |
| ZHAO S Q, PANG C Y, WEI H L, WANG H T, LI L B, SU J J, FAN S L, YU S X. Genetic inheritance of earliness traits in upland cotton (Gossypium hirsutum L.) inferred by joint analysis of multiple generations. Cotton Science, 2017, 29(2):119-127. (in Chinese) | |
| [6] | 喻树迅, 王寒涛, 魏恒玲, 宿俊吉. 棉花早熟性研究进展及其应用. 棉花学报, 2017, 29(S1):1-10. |
| YU S X, WANG H T, WEI H L, SU J J. Research progress and application of early maturity in upland cotton. Cotton Science, 2017, 29(S1):1-10. (in Chinese) | |
| [7] | 高玉千, 聂以春, 张献龙. 棉花抗黄萎病基因的QTL定位. 棉花学报, 2003, 15(2):73-78. |
| GAO Y Q, NIE Y C, ZHANG X L. QTL mapping of genes resistant to verticillium wilt in cotton. Cotton Science, 2003, 15(2):73-78. (in Chinese) | |
| [8] | 范术丽, 喻树迅, 宋美珍, 原日红. 短季棉早熟性的分子标记及QTL定位. 棉花学报, 2006, 18(3):135-139. |
| FAN S L, YU S X, SONG M Z, YUAN R H. Construction of molecular linkage map and QTL mapping for earliness in short season cotton. Cotton Science, 2006, 18(3):135-139. (in Chinese) | |
| [9] | 张西英, 李金荣, 朱永军, 韩璐, 张薇. 海岛棉(Gossypium barbadense L.)产量和早熟性状QTL定位. 植物遗传资源学报, 2012, 13(4):614-621. |
| ZHANG X Y, LI J R, ZHU Y J, HAN L, ZHANG W. QTL Mapping of yield and earliness-related traits in seaisland cotton (Gossypium barbadense L.). Journal of Plant Genetic Resources, 2012, 13(4):614-621. (in Chinese) | |
| [10] |
LI C Q, WANG X Y, DONG N, ZHAO H H, XIA Z, WANG R, RICHARD L C, WANG Q L. QTL analysis for early-maturing traits in cotton using two upland cotton (Gossypium hirsutum L.) crosses. Breeding Science, 2013, 63(2):154-163.
doi: 10.1270/jsbbs.63.154 |
| [11] |
JIA X Y, PANG C Y, WEI H L, WANG H T, MA Q F, YANG J L, CHENG S S, SU J J, FAN S L, SONG M Z, WUSIMAN N, YU S X. High-density linkage map construction and QTL analysis for earliness-related traits in Gossypium hirsutum L. BMC Genomics, 2016, 17(1):909.
doi: 10.1186/s12864-016-3269-y |
| [12] | 唐富福, 徐非非, 包劲松. 全基因组关联分析在水稻遗传育种中的应用. 核农学报, 2013, 27(5):598-606. |
| TANG F F, XU F F, BAO J S. Application of genome-wide association studies in rice genetics and breeding. Journal of Nuclear Agricultural Sciences, 2013, 27(5):598-606. (in Chinese) | |
| [13] |
ZHANG F, HU Z Q, WU Z C, LU J L, SHI Y Y, XU J L, WANG X Y, WANG J P, ZHANG F, WANG M M, SHI X R, CUI Y R, CASIANA V C, ZHUO D L, HU D D, LI M, WANG W S, ZHAO X Q, ZHENG T Q, FU B Y, JAUHAR A, ZHOU Y L, LI Z K. Reciprocal adaptation of rice and Xanthomonas oryzae pv. oryzae: Cross-species 2D GWAS reveals the underlying genetics. The Plant Cell, 2021, 33(8):2538-2561. doi: 10.1093/PLCELL/KOAB146.
doi: 10.1093/plcell/koab146 |
| [14] | 刘坤, 张雪海, 孙高阳, 闫鹏帅, 郭海平, 陈思远, 薛亚东, 郭战勇, 谢惠玲, 汤继华, 李卫华. 玉米株型相关性状的全基因组关联分析. 中国农业科学, 2018, 51(5):821-834. |
| LIU K, ZHANG X H, SUN G Y, YAN P S, GUO H P, CHEN S Y, XUE Y D, GUO Z Y, XIE H L, TANG J H, LI W H. Genome-wide association studies of plant type traits in maize. Scientia Agricultura Sinica, 2018, 51(5):821-834. (in Chinese) | |
| [15] | 张蕊, 邓文亚, 杨柳, 王亚萍, 肖芳枝, 禾健, 卢坤. 盐胁迫下甘蓝型油菜发芽期下胚轴和根长的全基因组关联分析. 中国农业科学, 2017, 50(1):15-35. |
| ZHANG R, DENG W Y, YANG L, WANG Y P, XIAO F Z, HE J, LU K. Genome-wide association study of root length and hypocotyl length at germination stage under saline conditions in Brassica napus. Scientia Agricultura Sinica, 2017, 50(1):15-35. (in Chinese) | |
| [16] | LIN F, SHABIR H W, PAUL J C, WEN Z X, LI W L, ZHANG N, AUSTIN G M, BI Y D, TAN R J, ZHANG S C, GU C H, MARTIN I C, WANG D C. QTL mapping and GWAS for identification of loci conferring partial resistance to pythium sylvaticum in soybean (Glycine max (L.) Merr). Molecular Breeding: New Strategies in Plant Improvement, 2020, 40(6):19-27. |
| [17] |
HUANG C, NIE X H, SHEN C, YOU C Y, LI W, ZHAO W X, LIN Z X. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs. Plant Biotechnology Journal, 2017, 15(11):1374-1386.
doi: 10.1111/pbi.2017.15.issue-11 |
| [18] |
LI L B, ZHANG C, HUANG J Q, LIU Q B, WEI H L, WANG H T, LIU G Y, GU L J, YU S X. Genomic analyses reveal the genetic basis of early maturity and identification of loci and candidate genes in upland cotton (Gossypium hirsutum L.). Plant Biotechnology Journal, 2021, 19(1):109-123.
doi: 10.1111/pbi.v19.1 |
| [19] |
SUN Z W, WANG X F, LIU Z W, GU Q S, ZHANG Y, LI Z K, KE H F, YANG J, WU J H, WU L Q, ZHANG GY, MA Z Y. Genome-wide association study discovered genetic variation and candidate genes of fiber quality traits in Gossypium hirsutum L. Plant Biotechnology Journal, 2017, 15(8):982-996.
doi: 10.1111/pbi.2017.15.issue-8 |
| [20] |
SUN Z W, WANG X F, LIU Z W, GU Q S, ZHANG Y, LI Z K, KE H F, YANG J, WU J H, WU L Q, ZHANG G Y, ZHANG C Y, MA Z Y. A genome-wide association study uncovers novel genomic regions and candidate genes of yield-related traits in upland cotton. Theoretical and Applied Genetics, 2018, 131(11):2413-2425.
doi: 10.1007/s00122-018-3162-y |
| [21] |
SHEN C, WANG N, HUANG C, WANG M J, ZHANG X L, LIN Z X. Population genomics reveals a fine-scale recombination landscape for genetic improvement of cotton. The Plant Journal, 2019, 99(3):494-505.
doi: 10.1111/tpj.v99.3 |
| [22] |
MA Z Y, HE S P, WANG X F, SUN J L, ZHANG Y, ZHANG G Y, WU L Q, LI Z K, LIU Z H, SUN G F, YAN Y Y, JIA Y H, YANG J, PAN Z E, GU Q S, LI X Y, SUN Z W, DAI P H, LIU Z W, GONG W F, WU J H, WANG M, LIU H W, FENG K Y, KE H F, WANG J D, LAN H Y, WANG G N, PENG J, WANG N, WANG L R, PANG B Y, PENG Z, LI R Q, DU X M. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nature Genetics, 2018, 50(6):803-813.
doi: 10.1038/s41588-018-0119-7 |
| [23] |
SU J J, WANG C X, YANG D L, SHI C H, ZHANG A, MA Q, LIU J J, ZHANG X L, HUANG L, MA X F. Decryption of favorable haplotypes and potential candidate genes for five fiber quality properties using a relatively novel genome-wide association study procedure in upland cotton. Industrial Crops and Products, 2020, 158:113004.
doi: 10.1016/j.indcrop.2020.113004 |
| [24] |
SU J J, PANG C Y, WEI H L, LI L B, LIANG B, WANG C X, SONG M Z, WANG H T, ZHAO S Q, JIA X Y, MAO G Z, HUANG L, GENG D D, WANG C S, YU S X. Identification of favorable SNP alleles and candidate genes for traits related to early maturity via GWAS in upland cotton. BMC Genomics, 2016, 17:687.
doi: 10.1186/s12864-016-2875-z |
| [25] |
LI C Q, WANG Y Y, AI N J, LI Y, SONG J F. A genome-wide association study of early-maturation traits in upland cotton based on the cottonSNP8oK array. Journal of Integrative Plant Biology, 2018, 60(10):970-985.
doi: 10.1111/jipb.v60.10 |
| [26] | 王香茹, 张恒恒, 胡莉婷, 庞念厂, 董强, 贵会平, 宋美珍, 张西岭. 新疆棉区棉花脱叶催熟剂的筛选研究. 中国棉花, 2018, 45(2):8-14. |
| WANG X R, ZHANG H H, HU L T, PANG N C, DONG Q, GUI H P, SONG M Z, ZHANG X L. Screening for suitable cotton harvest aids in Xinjiang. China Cotton, 2018, 45(2):8-14. (in Chinese) | |
| [27] | 高丽丽. 脱叶剂喷施时间对棉花生理调节效应的研究. 乌鲁木齐: 新疆农业大学, 2016. |
| GAO L L. Study of defoliants spraying time on cotton physiological mechanism. Urumqi: Xinjiang Agricultural University, 2016. (in Chinese) | |
| [28] | 李俊文, 贾菲, 孙福鼎, 刘爱英, 石玉真, 龚举武, 商海红, 王涛, 巩万奎, 贾新合, 张建宏, 袁有禄, 华金平. 陆地棉吐絮铃数及吐絮率的QTL定位. 棉花学报, 2013, 25(6):471-477. |
| LI J W, JIA F, SUN F D, LIU A Y, SHI Y Z, GONG J W, SHANG H H, WANG T, GONG W K, JIA X H, ZHANG J H, YUAN Y L, HUA J P. Quantitative trait locus mapping of number and percentage of cracked and open bolls in Gossypium hirsutum L. Cotton Science, 2013, 25(6):471-477. (in Chinese) | |
| [29] |
HE J B, MENG S, ZHAO T J, XING G N, YANG S P, LI Y, GUAN R Z, LU J J, WANG Y F, XIA Q J, YANG B, GAI J Y. An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theoretical and Applied Genetics, 2017, 130(11):2327-2343.
doi: 10.1007/s00122-017-2962-9 |
| [30] | 盖钧镒, 贺建波. 限制性两阶段多位点全基因组关联分析法(RTM-GWAS)的特点、常见提问与应用前景. 中国农业科学, 2020, 53(9):1699-1703. |
| GAI J Y, HE J B. Major characteristics, often-raised queries and potential usefulness of the restricted two-stage multi-locus genome- wide association analysis. Scientia Agricultura Sinica, 2020, 53(9):1699-1703. (in Chinese) | |
| [31] | 潘丽媛, 贺建波, 赵晋铭, 王吴彬, 邢光南, 喻德跃, 张小燕, 李春燕, 陈受宜, 盖钧镒. RTM-GWAS方法应用于大豆RIL群体百粒重QTL检测的功效. 中国农业科学, 2020, 53(9):1730-1742. |
| PAN L Y, HE J B, ZHAO J M, WANG W B, XING G N, YU D Y, ZHANG X Y, LI C Y, CHEN S Y, GAI J Y. Detection power of RTM-GWAS applied to 100-seed weight QTL identification in a recombinant inbred lines population of soybean. Scientia Agricultura Sinica, 2020, 53(9):1730-1742. (in Chinese) | |
| [32] |
ZHANG Y, HE J, WANG Y, XING G, ZHAO J, LI Y, YANG S, PALMER R G, ZHAO T, GAI J. Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes. Journal of Experimental Botany, 2015, 66(20):6311-6325.
doi: 10.1093/jxb/erv342 |
| [33] |
ZHANG Y H, HE J B, WANG H W, MENG S, XING G N, LI Y, YANG S P, ZHAO J M, ZHAO T J, GAI J Y. Detecting the QTL-allele system of seed oil traits using multi-locus genome-wide association analysis for population characterization and optimal cross prediction in soybean. Frontiers in Plant Science, 2018, 9:1793.
doi: 10.3389/fpls.2018.01793 |
| [34] | 李曙光, 曹永策, 贺建波, 王吴彬, 邢光南, 杨加银, 赵团结, 盖钧镒. 大豆巢式关联作图群体蛋白质含量的遗传解析. 中国农业科学, 2020, 53(9):1743-1755. |
| LI S G, CAO Y C, HE J B, WANG W B, XING G N, YANG J Y, ZHAO T J, GAI J Y. Genetic dissection of protein content in a nested association mapping population of soybean. Scientia Agricultura Sinica, 2020, 53(9):1743-1755. (in Chinese) | |
| [35] | 刘再东, 孟珊, 贺建波, 邢光南, 王吴彬, 赵团结, 盖钧镒. 大豆重组自交系群体异黄酮含量QTL连锁定位与关联定位的比较研究. 中国农业科学, 2020, 53(9):1756-1772. |
| LIU Z D, MENG S, HE J B, XING G N, WANG W B, ZHAO T J, GAI J Y. A comparative study on linkage and association QTL mapping for seed isoflavone contents in a recombinant inbred line population of soybean. Scientia Agricultura Sinica, 2020, 53(9):1756-1772. (in Chinese) | |
| [36] |
SU J J, WANG C X, MA Q, ZHANG A, SHI C H, LIU J J, ZHANG X L, YANG D L, MA X F. An RTM-GWAS procedure reveals the QTL alleles and candidate genes for three yield-related traits in upland cotton. BMC Plant Biology, 2020, 20(1):416-416.
doi: 10.1186/s12870-020-02613-y |
| [37] |
HU Y, CHEN J D, FANG L, ZHANG Z Y, MA W, NIU Y C, JU L Z, DENG J Q, ZHAO T, LIAN J M, KOBI B, FANG D, LIU X, RUAN Y L, RAHMAN M, HAN J L, WANG K, WANG Q, WU H T, MEI G F, ZANG Y H, HAN Z G, XU C Y, SHEN W J, YANG D F, SI Z F, DAI F, ZOU L F, HUANG F, BAI Y L, ZHANG Y G, AVITAL B, HILLA B H, ZHU X F, ZHOU B L, GUAN X Y, ZHU S J, CHEN X Y, ZHANG T Z. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nature Genetics, 2019, 51(4):739-748.
doi: 10.1038/s41588-019-0371-5 |
| [38] | 郝晓帅, 傅蒙蒙, 刘再东, 贺建波, 王燕平, 任海祥, 王德亮, 杨兴勇, 程延喜, 杜维广, 盖钧镒. 东北大豆种质群体百粒重QTL-等位变异的全基因组解析. 中国农业科学, 2020, 53(9):1717-1729. |
| HAO X S, FU M M, LIU Z D, HE J B, WANG Y P, REN H X, WANG D L, YANG X Y, CHENG Y X, DU W G, GAI J Y. Genome-Wide QTL-allele dissection of 100-seed weight in the northeast China soybean germplasm population. Scientia Agricultura Sinica, 2020, 53(9):1717-1729. (in Chinese) | |
| [39] | 马树庆, 杨菲芸 我国霜期、霜冻时空特征及其对气候变暖的响应. 气象灾害防御, 2015, 22(2):1-14+36. |
| MA S Q, YANG F Y. Temporal and spatial characteristics of frost period and frost and its response to climate warming in China. Meteorological Disaster Prevention, 2015, 22(2):1-14+36.(in Chinese) | |
| [40] | 王林, 张强, 马江锋, 朱玉永, 田英, 李红, 毕显杰, 宋敏, 王海标, 雷天翔, 李召虎, 田晓莉, 杜明伟, 张立祯, 赵冰梅. 新疆棉区植保无人机喷施棉花脱叶催熟剂效果研究. 棉花学报, 2021, 33(3):200-208. |
| WANG L, ZHANG Q, MA J F, ZHU Y Y, TIAN Y, LI H, BI X J, SONG M, WANG H B, LEI T X, LI Z H, TIAN X L, DU M W, ZHANG L Z, ZHAO B M. Study on the effect of spraying cotton defoliant by plant protection UAVs in Xinjiang cotton area. Cotton Science, 2021, 33(3):200-208. (in Chinese) | |
| [41] |
艾尼江, 刘任重, 赵图强, 秦江鸿, 张天真. 陆地棉早熟基因来源的遗传分析. 作物学报, 2013, 39(9):1548-1561.
doi: 10.3724/SP.J.1006.2013.01548 |
|
AI N J, LIU R Z, ZHAO T Q, QIN J H, ZHANG T Z. Analysis of early maturity gene sources in upland cotton using molecular markers. Acta Agronomica Sinica, 2013, 39(9):1548-1561. (in Chinese)
doi: 10.3724/SP.J.1006.2013.01548 |
|
| [42] | SU J J, FAN S L, LI L B, WEI H L, WANG C X, WANG H T, SONG M Z, ZHANG C, GU L J, ZHAO S Q, MAO G Z, WANG C S, PANG C Y, YU S X. Detection of favorable QTL alleles and candidate genes for lint percentage by GWAS in Chinese upland cotton. Frontiers in Plant Science, 2016, 7:1576. |
| [43] | 黄璐瑶. 水稻乙醇酸氧化酶GLO1,GLO4生理功能分析[D]. 长沙: 湖南农业大学, 2019. |
| HUANG L Y. The analysis of the physiological functions of glycolate oxidase GLO1 and GLO4 in rice[D]. Changsha: Hunan Agricultural University, 2019. (in Chinese) | |
| [44] |
PEI Z M, MURATA Y, BENNING G, THOMINE S, KLÜSENER B, ALLEN G J, GRILL E, SCHROEDER J I. Calcium channels activated by hydrogen peroxide mediate abscisic acid signalling in guard cells. Nature. 2000, 406(6797):731-4. doi: 10.1038/35021067.
doi: 10.1038/35021067 |
| [45] |
贺建波, 刘方东, 邢光南, 王吴彬, 赵团结, 管荣展, 盖钧镒. 限制性两阶段多位点全基因组关联分析方法的特点与计算程序. 作物学报, 2018, 44(9):1274-1289.
doi: 10.3724/SP.J.1006.2018.01274 |
|
HE J B, LIU D F, XING G N, WANG W B, ZHAO T J, GUAN R Z, GAI J Y. Characterization and analytical programs of the restricted two-stage multilocus genome-wide association analysis. Acta Agronomica Sinica, 2018, 44(9):1274-1289. (in Chinese)
doi: 10.3724/SP.J.1006.2018.01274 |
|
| [46] | 贺建波, 刘方东, 王吴彬, 邢光南, 管荣展, 盖钧镒. 限制性两阶段多位点全基因组关联分析法在遗传育种中的应用. 中国农业科学, 2020, 53(9):1704-1716. |
| HE J B, LIU F D, WANG W B, XING G N, GUAN R Z, GAI J Y. Restricted two-stage multi-locus genome-wide association analysis and its applications to genetic and breeding studies. Scientia Agricultura Sinica, 2020, 53(9):1704-1716. (in Chinese) |
| [1] | 王彩香,袁文敏,刘娟娟,谢晓宇,马麒,巨吉生,陈炟,王宁,冯克云,宿俊吉. 西北内陆早熟陆地棉品种的综合评价及育种演化[J]. 中国农业科学, 2023, 56(1): 1-16. |
| [2] | 胡盛,李阳阳,唐章林,李加纳,曲存民,刘列钊. 干旱胁迫下甘蓝型油菜籽粒含油量和蛋白质含量变化的全基因组关联分析[J]. 中国农业科学, 2023, 56(1): 17-30. |
| [3] | 职蕾,者理,孙楠楠,杨阳,Dauren Serikbay,贾汉忠,胡银岗,陈亮. 小麦苗期铅耐受性的全基因组关联分析[J]. 中国农业科学, 2022, 55(6): 1064-1081. |
| [4] | 王秀秀,邢爱双,杨茹,何守朴,贾银华,潘兆娥,王立如,杜雄明,宋宪亮. 陆地棉种质资源表型性状综合评价[J]. 中国农业科学, 2022, 55(6): 1082-1094. |
| [5] | 李恒,字向东,王会,熊燕,吕明杰,刘宇,蒋旭东. 基于全基因组重测序的山羊产羔数性状关键调控基因的筛选[J]. 中国农业科学, 2022, 55(23): 4753-4768. |
| [6] | 李婷,董远,张君,冯志前,王亚鹏,郝引川,张兴华,薛吉全,徐淑兔. 玉米杂交种穗部性状的全基因组关联分析[J]. 中国农业科学, 2022, 55(13): 2485-2499. |
| [7] | 王娟, 马晓梅, 周小凤, 王新, 田琴, 李成奇, 董承光. 棉花产量构成因素性状的全基因组关联分析[J]. 中国农业科学, 2022, 55(12): 2265-2277. |
| [8] | 刘瑞达, 葛常伟, 王敏轩, 申延会, 李朋珍, 崔子倩, 刘瑞华, 沈倩, 张思平, 刘绍东, 马慧娟, 陈静, 张桂寅, 庞朝友. 陆地棉转录因子基因GhMYB108的克隆及其在抗旱中的作用[J]. 中国农业科学, 2022, 55(10): 1877-1890. |
| [9] | 崔承齐, 刘艳阳, 江晓林, 孙知雨, 杜振伟, 武轲, 梅鸿献, 郑永战. 芝麻产量相关性状的多位点全基因组关联分析及候选基因预测[J]. 中国农业科学, 2022, 55(1): 219-232. |
| [10] | 袁景丽,郑红丽,梁先利,梅俊,余东亮,孙玉强,柯丽萍. 花青素代谢对陆地棉叶片和纤维色泽呈现的影响[J]. 中国农业科学, 2021, 54(9): 1846-1855. |
| [11] | 秦鸿德, 冯常辉, 张友昌, 别墅, 张教海, 夏松波, 王孝刚, 王琼珊, 蓝家样, 陈全求, 焦春海. 基于部分NCII设计的陆地棉F1表现预测[J]. 中国农业科学, 2021, 54(8): 1590-1598. |
| [12] | 张鹏飞,史良玉,刘家鑫,李洋,吴成斌,王立贤,赵福平. 畜禽全基因组长纯合片段检测的研究进展[J]. 中国农业科学, 2021, 54(24): 5316-5326. |
| [13] | 王娜,赵资博,高琼,何守朴,马晨辉,彭振,杜雄明. 陆地棉盐胁迫应答基因GhPEAMT1的克隆及功能分析[J]. 中国农业科学, 2021, 54(2): 248-260. |
| [14] | 严勇亮,时晓磊,张金波,耿洪伟,肖菁,路子峰,倪中福,丛花. 春小麦籽粒主要品质性状的全基因组关联分析[J]. 中国农业科学, 2021, 54(19): 4033-4047. |
| [15] | 宋春晖,陈晓菲,王枚阁,郑先波,宋尚伟,焦健,王苗苗,马锋旺,白团辉. 基于SLAF-seq技术鉴定苹果砧木耐涝候选基因[J]. 中国农业科学, 2021, 54(18): 3932-3944. |
|
||