






中国农业科学 ›› 2020, Vol. 53 ›› Issue (15): 3005-3019.doi: 10.3864/j.issn.0578-1752.2020.15.002
余爱丽( ),赵晋锋(),成锴,王振华,张鹏,刘鑫,田岗,赵太存,王玉文()
),赵晋锋(),成锴,王振华,张鹏,刘鑫,田岗,赵太存,王玉文()
收稿日期:2019-07-17
接受日期:2020-02-02
出版日期:2020-08-01
发布日期:2020-08-06
通讯作者:
王玉文
作者简介:余爱丽,E-mail: 基金资助:
YU AiLi(),ZHAO JinFeng(),CHENG Kai,WANG ZhenHua,ZHANG Peng,LIU Xin,TIAN Gang,ZHAO TaiCun,WANG YuWen()
Received:2019-07-17
Accepted:2020-02-02
Online:2020-08-01
Published:2020-08-06
Contact:
YuWen WANG
摘要:
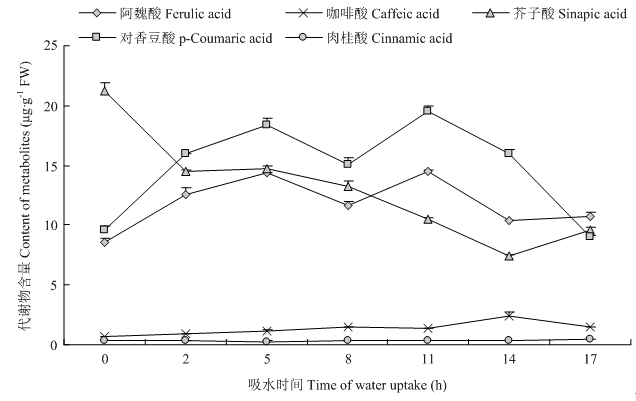
【目的】谷子适应性强,抗旱耐瘠,是起源于中国的重要作物。通过转录组测序技术分析谷子萌发不同吸水期的转录组差异,以期获得谷子萌发过程中的差异表达基因,寻找调控谷子萌发的重要代谢途径和代谢物。【方法】以晋谷20为材料,构建谷子萌发过程中开始快速吸水期、滞缓吸水期和重新大量吸水期的cDNA文库,进行转录组分析;采用K-Means开展基因表达聚类分析;利用DESeq筛选差异表达基因;通过COG、GO、KEGG等对差异表达基因进行功能注释;利用KEGG富集挖掘不同吸水期调控种子萌发的关键代谢途径和关键基因;并采用qRT-PCR验证其可靠性;用HPLC分析关键代谢物含量。【结果】转录物测序分析获得谷子萌发开始快速吸水期、滞缓吸水期和重新大量吸水期覆盖整个基因组的基因表达谱,共获得33 643个基因,识别9个具有不同表达模式的共表达基因簇。比较种子萌发的开始快速吸水期与滞缓吸水期、滞缓吸水期与重新大量吸水期、开始快速吸水期与重新大量吸水期,分别筛选出3 893、4 612和8 472个差异表达基因。KEGG富集分析表明,3个比较的差异表达基因都显著富集到phenylpropanoid biosynthesis、phenylalanine metabolism、starch and sucrose metabolism代谢途径;开始快速吸水期与滞缓吸水期、开始快速吸水期与重新大量吸水期的差异表达基因还显著富集到plant hormone signal transduction途径。并且3个比较中富集到phenylpropanoid biosynthesis和phenylalanine metabolism代谢途径的差异表达基因数都最多,其中过氧化物酶基因(peroxidase)比例最高。通过qRT-PCR对4个苯丙烷生物合成途径相关基因的分析表明,其表达趋势与转录组分析结果基本一致,其中,4-香豆酸-CoA连接酶3(4-coumarate-CoA ligase 3)在谷子种子中存在已形成mRNA,萌发吸水过程中呈先下调后上调再下调的表达趋势。苯丙烷类相关代谢物含量分析显示,芥子酸在种子中大量储备,在萌发过程中呈下调趋势;阿魏酸、对香豆酸和咖啡酸呈先上调后下调趋势。【结论】谷子萌发过程中,不同吸水期的差异表达基因显著与苯丙烷生物合成途径和苯丙氨酸代谢途径相关;其上游基因4-香豆酰-辅酶A连接酶和下游基因过氧化物酶家族成员在谷子萌发响应水分过程中发挥调控作用;中间产物芥子酸可能参与种子的休眠与萌发。
余爱丽,赵晋锋,成锴,王振华,张鹏,刘鑫,田岗,赵太存,王玉文. 谷子萌发吸水期关键代谢途径的筛选与分析[J]. 中国农业科学, 2020, 53(15): 3005-3019.
YU AiLi,ZHAO JinFeng,CHENG Kai,WANG ZhenHua,ZHANG Peng,LIU Xin,TIAN Gang,ZHAO TaiCun,WANG YuWen. Screening and Analysis of Key Metabolic Pathways in Foxtail Millet During Different Water Uptake Phases of Germination[J]. Scientia Agricultura Sinica, 2020, 53(15): 3005-3019.
表1
苯丙烷类相关基因qRT-PCR表达分析引物"
| 基因名称 Gene name | 基因ID Gene ID | 正向引物 Forward primer (5'-3') | 反向引物 Reverse primer (5'-3') |
|---|---|---|---|
| β-Actin | Seita.7G294000 | TGTGCCGGCCATGTATGT | CACACCATCACCAGAGTCCAA |
| Cationic peroxidase SPC4 precursor | Seita.3G004800 | CGGCATCGTCAGGGATTT | AGGCGGGCTTTGTTGGT |
| 4-coumarate--CoA ligase 3 | Seita.1G065800 | TGGAGTTCGCCGAGGTGAT | CGAGTTGAGCGAGTAGATGTGG |
| Beta-glucosidase 6 precursor | Seita.9G492600 | CCGGCGTGTCACTAATGCT | GTCTTCCCTCTCCCATCTTCCT |
| Peroxidase 4 precursor | Seita.5G145500 | TCTCGACGCTCCTGTCCAT | TTGGTGGCGGTGTCGTT |
表2
测序结果及其与参考基因组比对分析"
| 分类 Classification | 开始快速吸水期PhaseⅠ | 滞缓吸水期PhaseⅡ | 重新大量吸水期PhaseⅢ | |||
|---|---|---|---|---|---|---|
| CK2 | CK2R | CK8 | CK8R | CK14 | CK14R | |
| 总reads Clean reads | 38845760 | 39679812 | 56164700 | 43525742 | 46838670 | 49151796 |
| 总核苷酸 Clean bases | 4894565760 | 4986871932 | 7076752200 | 5472201146 | 5901672420 | 6178141042 |
| GC含量 GC content (%) | 58.72 | 57.83 | 57.53 | 56.81 | 57.21 | 56.36 |
| 碱基质量值 Q30 (%) | 89.74 | 85.29 | 90.33 | 85.17 | 91.62 | 85.13 |
| 匹配的reads Mapped reads | 31007621 (79.82%) | 31533133 (79.47%) | 45812237 (81.57%) | 35213766 (80.90%) | 38024006 (81.18%) | 39595878 (80.56%) |
| 唯一位点匹配reads Unique mapped reads | 26519605 (68.27%) | 27126368 (68.36%) | 42124995 (75.00%) | 31824703 (73.12%) | 35017461 (74.76%) | 35922812 (73.09%) |
| 多位点匹配reads Multiple mapped reads | 4488016 (11.55%) | 4406765 (11.11%) | 3687242 (6.57%) | 3389063 (7.79%) | 3006545 (6.42%) | 3673066 (7.47%) |
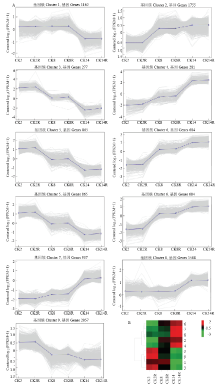
图1
基因表达谱K-Means聚类图 A:具有不同表达模式的9个基因簇,纵坐标表示中心化的基因表达量的对数值;B:9个基因簇基因表达热图"


图2
差异表达基因韦恩图 A:所有差异表达基因;B:上调差异表达基因;C:下调差异表达基因"
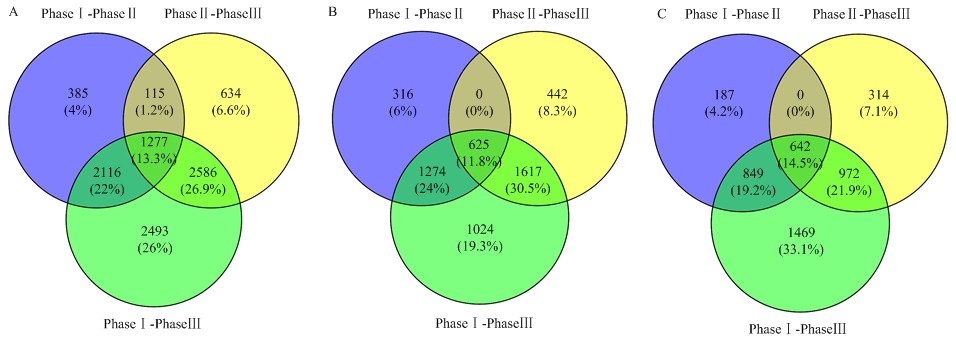

图3
差异表达基因火山图 横坐标表示某一个基因在两样品中表达量差异倍数的对数值;纵坐标表示基因表达量变化的统计学显著性的负对数值"
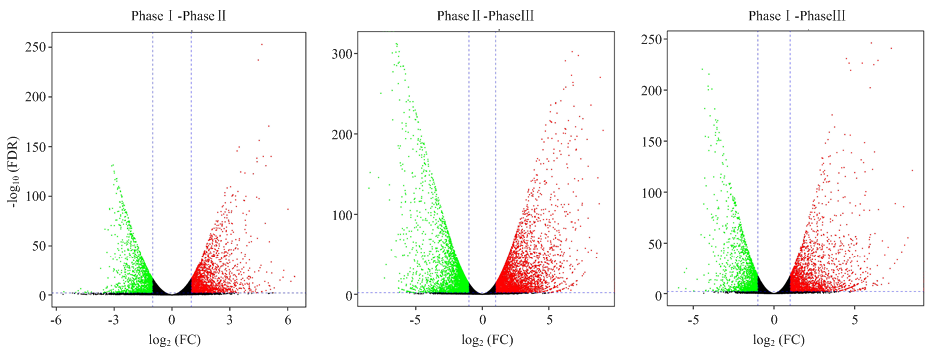
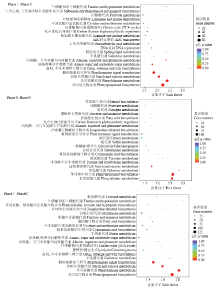
图4
差异表达基因KEGG通路富集图 横坐标为富集因子(Rich Factor),表示差异表达基因中注释到某通路的基因比例与所有基因中注释到该通路的基因比例的比值。纵坐标表示通路名称;圆圈的大小表示通路中富集的基因数目,圆圈越大,表示基因越多;圆圈的颜色代表q value"
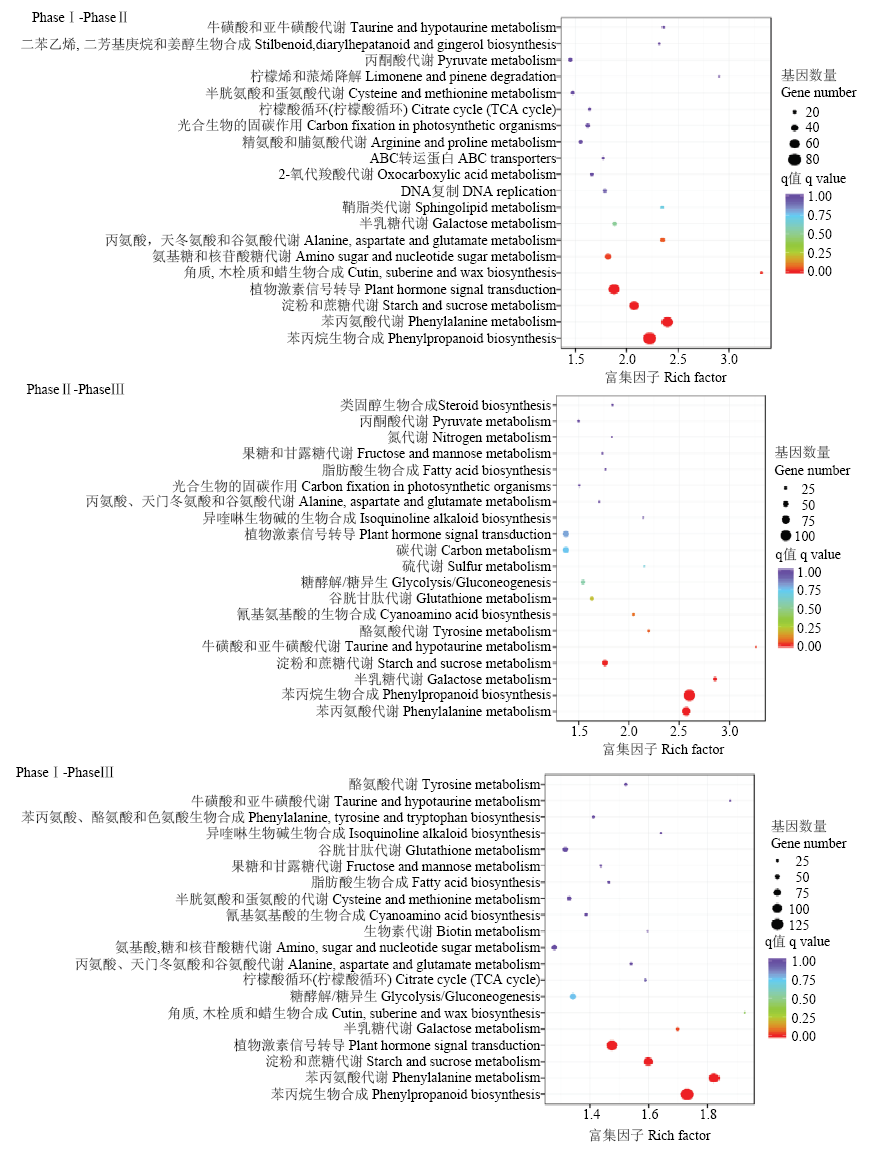
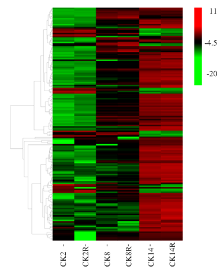
图5
苯丙烷类相关的不同萌发吸水阶段间差异表达基因的表达分析"
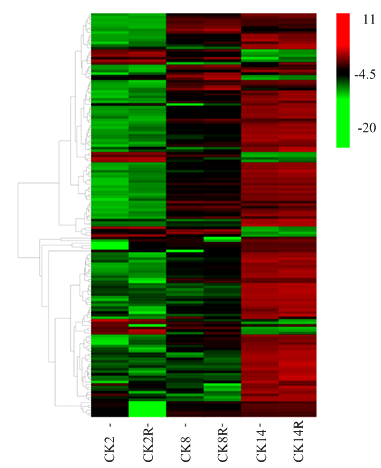
图6
苯丙烷类相关的不同萌发吸水阶段间差异表达基因的功能分类"

图7
苯丙烷类相关基因在不同萌发吸水时期的qRT-PCR表达分析"
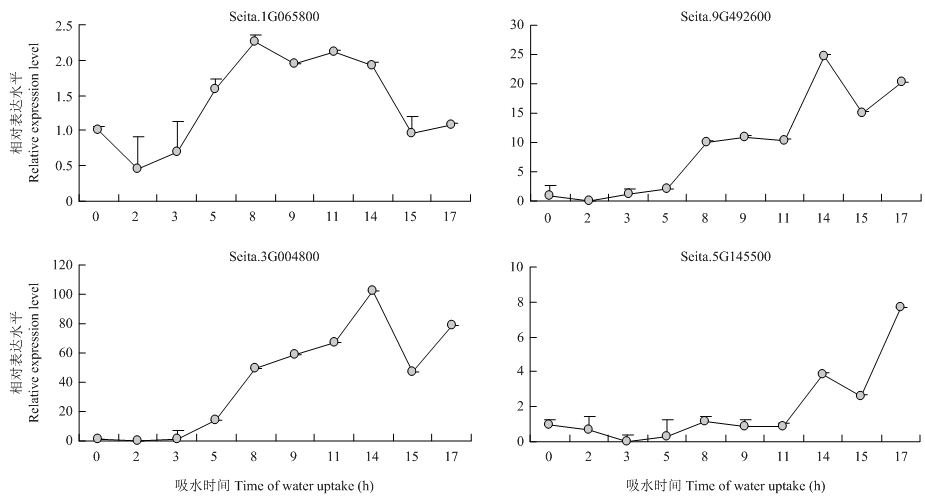
图8
苯丙烷类相关代谢物不同萌发吸水时期含量变化"
| [1] | BEWLEY J D, BLACK M. Seeds: Physiology of Development and Germination. New York: Plenum Press, 1994. |
| [2] | 毕辛华, 戴心维. 种子学. 北京: 农业出版社, 1993. |
| BI X H, DAI X W. Seed Science Beijing: Agricultural Press, 1993. (in Chinese) | |
| [3] |
BEWLEY J D. Seed germination and dormancy. The Plant Cell, 1997,9:1055-1066.
doi: 10.1105/tpc.9.7.1055 pmid: 12237375 |
| [4] | 张锦鹏, 王茅雁, 白云凤, 贾晋平, 王国英. 谷子品种抗旱性的苗期快速鉴定. 植物遗传资源学报, 2005(1):59-62. |
| ZHANG J P, WANG M Y, BAI Y F, JIA J P, WANG G Y. Rapid evaluation on drought tolerance of foxtail millet at seedling stage. Journal of Plant Genetic Resources, 2005(1):59-62. (in Chinese) | |
| [5] | 朱学海. 谷子耐旱资源筛选及其遗传多样性分析[D]. 北京: 中国农业科学院, 2008. |
| ZHU X H. Selecting drought resources and its analysis of genetic diversity in foxtail millet[D]. Beijing: Chinese Academy of Agricultural Sciences, 2008. (in Chinese) | |
| [6] | 秦岭, 杨延兵, 管延安, 张华文, 王海莲, 刘宾, 陈二影. 不同生态区主要育成谷子品种芽期耐旱性鉴定. 植物遗传资源学报, 2013,14(1):146-151. |
| QIN L, YANG Y B, GUAN Y A, ZHANG H W, WANG H L, LIU B, CHEN E Y. Identification of drought tolerance at germination period of foxtail millet cultivars developed from different ecological regions. Journal of Plant Genetic Resources, 2013,14(1):146-151. (in Chinese) | |
| [7] | BHEEMESH S J, SUBBA RAO M, REDDI SEKHAR M, LATHA P. Effect of PEG-induced drought stress on germination early seedling growth of foxtail millet varieties. International Journal of Current Microbiology and Applied Sciences, 2018, special issue-6:2674-2681. |
| [8] | 王春梅, 闫双堆, 卜玉山, 刘利军, 张乃于. As对谷子萌发、幼苗生长及抗氧化酶系统的影响. 水土保持学报, 2018,32(6):354-360. |
| WANG C M, YAN S D, BU Y S, LIU L J, ZHANG N Y. Effect of As stress on millet germination, seedling growth and antioxidant system. Journal of Soil and Water Conservation, 2018,32(6):354-360. (in Chinese) | |
| [9] |
裴帅帅, 尹美强, 温银元, 黄明镜, 张彬, 郭平毅, 王玉国, 原向阳. 不同品种谷子种子萌发期对干旱胁迫的生理响应及其抗旱性评价. 核农学报, 2014,28(10):1897-1904.
doi: 10.11869/j.issn.100-8551.2014.10.1897 |
|
PEI S S, YIN M Q, WEN Y Y, HUANG M J, ZHANG B, GUO P Y, WANG Y G, YUAN X Y. Physiological response of foxtail millet to drought stress during seed germination and drought resistance evaluation. Journal of Nuclear Agricultural Sciences, 2014,28(10):1897-1904. (in Chinese)
doi: 10.11869/j.issn.100-8551.2014.10.1897 |
|
| [10] | 杨净, 王宏富, 鱼冰星, 李智, 王钰云. 超重力对谷子种子萌发及生理生化特性的影响. 激光生物学报, 2018,27(4):83-88. |
| YANG J, WANG H F, YU B X, LI Z, WANG Y Y. Effects of hypergravity on seed germination and physiological and biochemical characteristics of foxtail millet. Acta Laser Biology Sinica, 2018,27(4):83-88. (in Chinese) | |
| [11] |
TANG S, LI L, WANG Y, CHEN Q, ZHANG W, JIA G, ZHI H, ZHAO B, DIAO X. Genotype-specific physiological and transcriptomic responses to drought stress in Setaria italica (an emerging model for Panicoideae grasses). Scientific Reports, 2017,7(1):10009.
pmid: 28855520 |
| [12] | 窦祎凝, 秦玉海, 闵东红, 张小红, 王二辉, 刁现民, 贾冠清, 徐兆师, 李连城, 马有志, 陈明. 谷子转录因子SiNAC18通过ABA信号途径正向调控干旱条件下的种子萌发. 中国农业科学, 2017,50(16):3071-3081. |
| DOU Y N, QIN Y H, MIN D H, ZHANG X H, WANG E H, DIAO X M, JIA G Q, XU Z S, LI L C, MA Y Z, CHEN M. Transcription factor SiNAC18 positively regulates seed germination under drought stress through ABA signaling pathway in foxtail millet (Setaria italic L.). Scientia Agricultura Sinica, 2017,50(16):3071-3081. (in Chinese) | |
| [13] |
许冰霞, 尹美强, 温银元, 裴帅帅, 柯贞进, 张彬, 原向阳. 谷子萌发期响应干旱胁迫的基因表达谱分析. 中国农业科学, 2018,51(8):1431-1447.
doi: 10.3864/j.issn.0578-1752.2018.08.002 |
|
XU B X, YIN M Q, WEN Y Y, PEI S S, KE Z J, ZHANG B, YUAN X Y. Gene expression profiling of foxtail millet (Setaria italica L.) under drought stress during germination. Scientia Agricultura Sinica, 2018,51(8):1431-1447. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2018.08.002 |
|
| [14] |
潘教文, 李臻, 王庆国, 管延安, 李小波, 戴绍军, 丁国华, 刘炜. NaCl处理谷子萌发期种子的转录组学分析. 中国农业科学, 2019,52(22):3964-3975.
doi: 10.3864/j.issn.0578-1752.2019.22.003 |
|
PAN J W, LI Z, WANG Q G, GUAN Y A, LI X B, DAI S J, DING G H, LIU W. Transcriptomics analysis of NaCl response in foxtail millet (Setaria italica L.) seeds at germination stage. Scientia Agricultura Sinica, 2019,52(22):3964-3975. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2019.22.003 |
|
| [15] | WANG X, WANG L J, YAN X C, WANG L, TAN M L, GENG X X, WEI W H. Transcriptome analysis of the germinated seeds identifies low-temperature responsive genes involved in germination process in Ricinus communis. Acta Physiologiae Plantarum, 2016,38(1):6. |
| [16] | DIXON R A. Stress-induced phenylpropanoid metabolism. The Plant Cell Online, 1995,7:1085-1097. |
| [17] |
LA CAMERA S, GOUZERH G, DHONDT S, HOFFMANN L, FRITIG B, LEGRAND M, HEITZ T. Metabolic reprogramming in plant innate immunity: The contributions of phenylpropanoid and oxylipin pathways. Immunological Reviews, 2004,198:267-284.
pmid: 15199968 |
| [18] |
VOGT T. Phenylpropanoid biosynthesis. Molecular Plant, 2010,3:2-20.
pmid: 20035037 |
| [19] | LIU X Y, WANG P P, WU Y F, CHENG A X, LOU H X. Cloning and functional characterization of two 4-coumarate: CoA ligase genes from Selaginella moellendorffii. Molecules, 2018,23:595. |
| [20] | HAHLBROCK K, SCHEEL D. Physiology and molecular biology of phenylpropanoid metabolism. Annual Review of Plant Physiology and Plant Molecular Biology, 1989,40:347-369. |
| [21] |
KIM D S, HWANG B K. An important role of the pepper phenylalanine ammonia-lyase gene (PAL1) in salicylic acid-dependent signalling of the defence response to microbial pathogens. Journal of Experimental Botany, 2014,65(9):2295-2306.
pmid: 24642849 |
| [22] |
KIM D, PERTEA G, TRAPNELL C, PIMENTEL H, KELLEY R, SALZBERG S L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 2013,14:R36.
doi: 10.1186/gb-2013-14-4-r36 pmid: 23618408 |
| [23] |
FLOREA L, SONG L, SALZBERG S L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research, 2013,2:188.
doi: 10.12688/f1000research.2-188.v2 pmid: 24555089 |
| [24] | SCHULZE S K, KANWAR R, GÖLZENLEUCHTER M, THERNEAU T M, BEUTLER A S. SERE: Single-parameter quality control and sample comparison for RNA-Seq. BMC genomics, 2012,13(1):524. |
| [25] |
ANDERS S, HUBER W. Differential expression analysis for sequence count data. Genome Biology, 2010,11(10):R106.
doi: 10.1186/gb-2010-11-10-r106 pmid: 20979621 |
| [26] | 邓泱泱, 荔建琦, 吴松锋, 朱云平, 陈耀文, 贺福初. nr数据库分析及其本地化. 计算机工程, 2006,32(5):71-74. |
| DENG Y Y, LI J Q, WU S F, ZHU Y P, CHEN Y W, HE F C. Integrated nr database in protein annotation system and its localization. Computer Engineering, 2006,32(5):71-74. (in Chinese) | |
| [27] | APWEILER R, BAIROCH A, WU C H, BARKER W C, YEH L S L. Uniprot: The universal protein knowledgebase. Nucleic Acids Research, 2004,32(D1):D115-D119. |
| [28] | KANEHISA M, GOTO S, KAWASHIMA S, OKUNO Y, HATTORI M. The KEGG resource for deciphering the genome. Nucleic Acids Research, 2004(32):D277-D280. |
| [29] | TATUS R L, GALPERIN M Y, NATALE D A, KOONIN E V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Reserch, 2000,28(1):33-36. |
| [30] | ASHBURNER M, BALL C A, BLAKE J A, BOTSTEIN D, BUTLER H, CHERRY J M, DAVIS A P, DOLINSKI K, DWIGHT S S, EPPIG J T, HARRIS M A, HILL D P, ISSEL-TARVER L, KASARSKIS A, LEWIS S, MATESE J C, RICHARDSON J E, RINGWALD M, RUBIN G M, SHERLOCK G. Gene ontology: Tool for the unification of biology. Gene, 2000,25(1):25-29. |
| [31] | ALEXA A, RAHNENFUHRER J. topGO: Enrichment analysis for gene ontology.R package version 2..8, 2010. |
| [32] |
SUPEK F, BOŠNJAK M, ŠKUNCA N, ŠMUC T. REVIGO summarizes and visualizes long lists of Gene Ontology terms. PLoS ONE, 2011,6:e21800
doi: 10.1371/journal.pone.0021800 pmid: 21789182 |
| [33] | PFAFFL M W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research, 2001,29:900. |
| [34] |
MOLNÁR-PERL I, FÜZFAI Z. Chromatographic, capillary electrophoretic and capillary electrochromatographic techniques in the analysis of flavonoids. Journal of Chromatography A, 2005,1073:201-227.
doi: 10.1016/j.chroma.2004.10.068 |
| [35] |
CATUSSE J, JOB C, JOB D. Transcriptome- and proteome-wide analyses of seed germination. Comptes Rendus Biologies, 2008,331:815-822.
doi: 10.1016/j.crvi.2008.07.023 pmid: 18926496 |
| [36] |
RAJJOU L, DUVAL M, GALLARDO K, CATUSSE J, BALLY J, JOB C, JOB D. Seed germination and vigor. Annual Review of Plant Biology, 2012,63:507-533.
doi: 10.1146/annurev-arplant-042811-105550 pmid: 22136565 |
| [37] |
FERRER J L, AUSTIN M B, STEWART C J R, NOEL J P. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiology and Biochemistry, 2008,46:356-370.
pmid: 18272377 |
| [38] |
REIGOSA M J, MALVIDO-PAZOS E. Phytotoxic effects of 21 plant secondary metabolites on Arabidopsis thaliana germination and root growth. Journal of Chemical Ecology, 2007,33:1456-1466.
doi: 10.1007/s10886-007-9318-x pmid: 17577597 |
| [39] | EDWARDS K, CRAMER C L, BOLWELL G P, DIXON R A, SCHUCH W, LAMB C J. Rapid transient induction of phenylalanine ammonia-lyase mRNA in elicitor-treated bean cells. Proceedings of the National Academy of Sciences of the USA, 1985,82:6731-6735. |
| [40] |
LIANG X W, DRON M, CRAMER C L, DIXON R A, LAMB C J. Differential regulation of phenylalanine ammonia-lyase genes during plant development and by environmental cues. Journal of Biological Chemistry, 1989,264:14486-14492.
pmid: 2760071 |
| [41] |
LIANG X W, DRON M, SCHMID J, DIXON R A, LAMB C J. Developmental and environmental regulation of a phenyl- alanine ammonia-lyase-β-glucuronidase gene fusion in transgenic tobacco plants. Proceedings of the National Academy of Sciences of the USA, 1989,86:9284-9288.
doi: 10.1073/pnas.86.23.9284 pmid: 2594769 |
| [42] |
HUANG J, GU M, LAI Z, FAN B, SHI K, ZHOU Y H, YU J Q, CHEN Z. Functional analysis of the Arabidopsis PAL gene family in plant growth, development, and response to environmental stress. Plant Physiology, 2010,153:1526-1538.
doi: 10.1104/pp.110.157370 pmid: 20566705 |
| [43] |
PAYYAVULA R S, NAVARRE D A, KUHL J C, PANTOJA A, PILLAI S S. Differential effects of environment on potato phenylpropanoid and carotenoid expression. BMC Plant Biology, 2012,12:39.
doi: 10.1186/1471-2229-12-39 pmid: 22429339 |
| [44] |
JIN Q, YAO Y, CAI Y, LIN Y. Molecular cloning and sequence analysis of a phenylalanine ammonia-lyase gene from Dendrobium. PLoS ONE, 2013,8:e62352.
doi: 10.1371/journal.pone.0062352 pmid: 23638048 |
| [45] |
MACDONALD M J, D'CUNHA G B. A modern view of phenylalanine ammonia lyase. Biochemistry and Cell Biology, 2007,85:273-282.
pmid: 17612622 |
| [46] |
MAUCH-MANI B, SLUSARENKO A J. Production of salicylic acid precursors is a major function of phenylalanine mmonia-lyase in the resistance of Arabidopsis to Peronospora parasitica. The Plant Cell, 1996,8:203-212.
doi: 10.1105/tpc.8.2.203 pmid: 12239383 |
| [47] | NUGROHO L H, VERBERNE M C, VERPOORTE R. Activities of enzymes involved in the phenylpropanoid pathway in constitutively salicylic acid-producing tobacco plants. Plant Physiology and Biochemistry, 2002,40:775-760. |
| [48] |
CHAMAN M E, COPAJA S V, ARGANDONA V H. Relationships between salicylic acid content, phenylalanine ammonia-lyase (PAL) activity, and resistance of barley to aphid infestation. Journal of Agricultural and Food Chemistry, 2003,51:2227-2231.
doi: 10.1021/jf020953b pmid: 12670161 |
| [49] |
PASSARDI F, LONGET D, PENEL C, AND DUNAND C. The class III peroxidase multigenic in land plants family in rice and its evolution. Phytochemistry, 2004,65:1879-1893.
doi: 10.1016/j.phytochem.2004.06.023 pmid: 15279994 |
| [50] |
PASSARDI F, COSIO C, PENEL C, AND DUNAND C. Peroxidases have more functions than a Swiss army knife. Plant Cell Reports, 2005,24:255-265.
doi: 10.1007/s00299-005-0972-6 |
| [51] |
HIRAGA S, SASAKI K, ITO H, OHASHI Y, MATSUI H. A large family of class Ⅲ plant peroxidases. Plant Cell Physiology, 2001,42(5):462-468.
doi: 10.1093/pcp/pce061 pmid: 11382811 |
| [52] |
ALMAGRO L, GÓMEZ ROS L V, BELCHI-NAVARRO S, BRU R, ROS BARCELÓ A, PEDREŇO M A. Class III peroxidases in plant defence reactions. Journal of Experimental Botany, 2009,60(2):377-390.
doi: 10.1093/jxb/ern277 pmid: 19073963 |
| [53] |
HILAIRE E, YOUNG S A, WILLARD L H, MCGEE J D, SWEAT T, CHITTOOR J M, GUIKEMA J A, LEACH J E. Vascular defense responses in rice: Peroxidase accumulation in xylem parenchyma cells and xylem wall thickening. Molecular Plant-Microbe Interactions, 2001,14(12):1411-1419.
doi: 10.1094/MPMI.2001.14.12.1411 pmid: 11768536 |
| [54] |
COEGO A, RAMIREZ V, ELLUL P, MAYDA E, VERA P. The H2O2-regulated Ep5C gene encodes a peroxidase required for bacterial speck susceptibility in tomato. The Plant Journal, 2005,42(2):283-293.
doi: 10.1111/j.1365-313X.2005.02372.x pmid: 15807789 |
| [55] |
WELINDER K G. Superfamily of plant, fungal and bacterial peroxidases. Current Opinion in Structural Biology, 1992,2:388-393.
doi: 10.1016/0959-440X(92)90230-5 |
| [56] |
WELINDER K G, JUSTESEN A F, KJÆRSGÅRD I V H, JENSEN R B, RASMUSSEN S K, JEPERSEN H M, DUROUX L. Structural diversity and transcription of class III peroxidases from Arabidopsis thaliana. European Journal of Biochemistry, 2002,269:6063-6081.
doi: 10.1046/j.1432-1033.2002.03311.x |
| [57] |
GABALDÓN C, LÓPEZ-SERRANO M, PEDREŇO M A, ROS BARCELÓ A. Cloning and molecular characterization of the basic peroxidase isoenzyme from Zinnia elegans, an enzyme involves in lignin biosynthesis. Plant Physiology, 2005,139:1138-1154.
doi: 10.1104/pp.105.069674 pmid: 16258008 |
| [58] |
PASSARDI F, TOGNOLLI M, DE MEYER M, PENEL C, DUNAND C. Two cell wall associated peroxidases from Arabidopsis influence root elongation. Planta, 2006,223:965-974.
doi: 10.1007/s00425-005-0153-4 pmid: 16284776 |
| [59] |
COSTA, M M, HILLIOU F, DUARTE P, PEREIRA L G, ALMEIDA I, LEECH M, MEMELINK J, BARCELÓ A R, SOTTOMAYOR M. Molecular cloning and characterization of a vacuolar class III peroxidase involved in the metabolism of anticancer alkaloids in Catharanthus roseus. Plant Physiology, 2008,146:403-417.
doi: 10.1104/pp.107.107060 pmid: 18065566 |
| [60] |
MEI W, QIN Y, SONG W, LI J, ZHU Y. Cotton GhPOX1 encoding plant class III peroxidase may be responsible for the high level of reactive oxygen species production that is related to cotton fiber elongation. Journal of Genetics and Genomics, 2009,36:141-150.
doi: 10.1016/S1673-8527(08)60101-0 pmid: 19302970 |
| [1] | 贾冠清, 刁现民. 中国谷子种业创新现状与未来展望[J]. 中国农业科学, 2022, 55(4): 653-665. |
| [2] | 郭淑青,宋慧,柴少华,郭岩,石兴,杜丽红,邢璐,解慧芳,张扬,李龙,冯佰利,刘金荣,杨璞. 谷子生育期及穗相关性状的QTL定位[J]. 中国农业科学, 2022, 55(15): 2883-2898. |
| [3] | 武翠卿,孙静鑫,郭平毅,王宏富,武新慧. 农艺措施对谷子产量及抗倒伏力学性能的影响[J]. 中国农业科学, 2021, 54(6): 1127-1142. |
| [4] | 张婷,王根平,罗焱杰,李琳,高翔,程汝宏,师志刚,董立,张喜瑞,杨伟红,许立闪. 色差分析在优质小米选育中的应用[J]. 中国农业科学, 2021, 54(5): 901-908. |
| [5] | 李顺国, 刘斐, 刘猛, 程汝宏, 夏恩君, 刁现民. 中国谷子产业和种业发展现状与未来展望[J]. 中国农业科学, 2021, 54(3): 459-470. |
| [6] | 郭淑青,宋慧,杨清华,高金锋,高小丽,冯佰利,杨璞. 谷子株高及穗部性状主基因+多基因混合遗传模型分析[J]. 中国农业科学, 2021, 54(24): 5177-5193. |
| [7] | 张硕,智慧,唐婵娟,罗明昭,汤沙,贾冠清,贾彦超,刁现民. 谷子条纹叶突变体A36-S的细胞学特性分析及基因定位[J]. 中国农业科学, 2021, 54(14): 2952-2964. |
| [8] | 张林林,智慧,汤沙,张仁梁,张伟,贾冠清,刁现民. 谷子抽穗时间基因SiTOC1的表达与单倍型变异分析[J]. 中国农业科学, 2021, 54(11): 2273-2286. |
| [9] | 杨延兵,秦岭,王润丰,陈二影,尹秀波,刘玉芹,张素梅,丛新军,李国瑜,王乐政,管延安. 山东省不同生态条件气候因素对谷子产量的影响[J]. 中国农业科学, 2020, 53(7): 1348-1358. |
| [10] | 赵娟,尹艺臻,王晓璐,马春英,尹美强,温银元,宋喜娥,董淑琦,杨雪芳,原向阳. 不同品种谷子愈伤组织对拿捕净胁迫的生理响应[J]. 中国农业科学, 2020, 53(5): 917-928. |
| [11] | 郭美俊,白亚青,高鹏,申洁,董淑琦,原向阳,郭平毅. 二甲四氯胁迫对谷子幼苗叶片衰老特性和 内源激素含量的影响[J]. 中国农业科学, 2020, 53(3): 513-526. |
| [12] | 李会霞,田岗,王玉文,刘鑫,刘红. 谷子杂交种与亲本性状的遗传相关性[J]. 中国农业科学, 2020, 53(2): 239-246. |
| [13] | 常国蓉,李任建,张琦,张育铭,韩渊怀,张宝俊. 利用WGCNA鉴定谷子内源脱落酸响应禾生指梗霉胁迫的共表达基因[J]. 中国农业科学, 2020, 53(16): 3280-3293. |
| [14] | 刘思辰,曹晓宁,温琪汾,王海岗,田翔,王君杰,陈凌,秦慧彬,王纶,乔治军. 山西谷子地方品种农艺性状和品质性状的综合评价[J]. 中国农业科学, 2020, 53(11): 2137-2148. |
| [15] | 李剑峰,张博,全建章,王永芳,张小梅,赵渊,袁玺垒,贾小平,董志平. 基于SSR标记的谷子主要农艺性状关联位点检测及等位变异分析[J]. 中国农业科学, 2019, 52(24): 4453-4469. |
|
||