






中国农业科学 ›› 2023, Vol. 56 ›› Issue (6): 1086-1101.doi: 10.3864/j.issn.0578-1752.2023.06.006
渠清1,2( ), 刘宁2,3, 邹金鹏2,3, 张雅璇3, 贾慧3, 孙蔓莉2,3, 曹志艳2,3(), 董金皋2,3()
), 刘宁2,3, 邹金鹏2,3, 张雅璇3, 贾慧3, 孙蔓莉2,3, 曹志艳2,3(), 董金皋2,3()
收稿日期:2022-10-27
接受日期:2022-11-23
出版日期:2023-03-16
发布日期:2023-03-23
联系方式:
渠清,E-mail:qu_qing@126.com。
基金资助:
QU Qing1,2(), LIU Ning2,3, ZOU JinPeng2,3, ZHANG YaXuan3, JIA Hui3, SUN ManLi2,3, CAO ZhiYan2,3(), DONG JinGao2,3()
Received:2022-10-27
Accepted:2022-11-23
Published:2023-03-16
Online:2023-03-23
摘要:
【目的】拟轮枝镰孢(Fusarium verticillioides)引起的玉米穗腐病是我国玉米产区发生严重的病害之一,论文旨在了解病原菌与玉米籽粒互作过程中的基因表达差异,为揭示病原菌的致病机制和玉米抗病机制提供依据。【方法】对拟轮枝镰孢侵染玉米籽粒0、4、12和72 h的样品进行转录组测序,之后采用生物信息学分析,分别以玉米和拟轮枝镰孢基因组为参考,以|log2FC|≥1,P-adjust<0.05为阈值筛选互作过程中玉米和拟轮枝镰孢的差异表达基因,利用GO和KEGG对其进行功能注释及富集分析。Goatools软件分析植物-病原互作、MAPK途径和植物激素信号转导通路相关差异基因的表达变化,采用实时荧光定量PCR(qRT-PCR)方法对测序差异基因进行验证。【结果】在互作4、12和72 h后拟轮枝镰孢分别有140、400和1 945个基因上调表达,有9、302和1 784个基因下调表达;玉米分别有293、692 和1 426个基因上调表达,320、482和153个基因下调表达。GO和KEGG富集分析显示,侵染早期拟轮枝镰孢在细胞间隙生长,差异基因主要富集在RNA生物合成、细胞壁结构成分、脂肪酸生物合成、蛋白质代谢、碳水化合物代谢、生物过程和代谢过程等通路中。拟轮枝镰孢早期侵染触发了玉米活性氧(ROS)爆发,差异基因主要富集在对活性氧、过氧化氢的反应,几丁质酶、单加氧酶活性,木质素代谢过程等相关通路中。侵染后期拟轮枝镰孢继续在籽粒中定殖及扩展,差异基因主要富集在碳水化合物和细胞壁多糖分解代谢过程、跨膜转运、氧化还原酶活性等功能通路中。玉米主要通过苯丙素、木质素、类黄酮生物合成,MAPK 信号通路、植物-病原互作和植物激素信号转导等途径差异基因大量表达响应病原菌侵染。随机选取6个玉米和6个拟轮枝镰孢的差异表达基因进行qRT-PCR分析,基因表达规律与转录组测序结果一致,证实了RNA-seq的准确性。【结论】在病原菌侵染早期,拟轮枝镰孢在细胞间隙生长,触发玉米活性氧爆发,相关通路差异基因表达;侵染中后期,病原菌以淀粉为营养素,继续在籽粒中定殖及扩展,玉米通过苯丙素、木质素及几丁质酶的生物合成等方面的相关基因表达响应拟轮枝镰孢侵染,同时植物-病原互作、MAPK途径和激素信号转导等途径参与抗拟轮枝镰孢侵染。
渠清, 刘宁, 邹金鹏, 张雅璇, 贾慧, 孙蔓莉, 曹志艳, 董金皋. 拟轮枝镰孢与玉米籽粒互作的差异基因筛选及代谢通路分析[J]. 中国农业科学, 2023, 56(6): 1086-1101.
QU Qing, LIU Ning, ZOU JinPeng, ZHANG YaXuan, JIA Hui, SUN ManLi, CAO ZhiYan, DONG JinGao. Screening of Differential Genes and Analysis of Metabolic Pathways in the Interaction Between Fusarium verticillioides and Maize Kernels[J]. Scientia Agricultura Sinica, 2023, 56(6): 1086-1101.
表1
本研究所用实时荧光定量 PCR引物信息"
| 引物名称 Primer name | 引物序列 Primer sequence (5′- 3′) |
|---|---|
| Zm00001d026018-F | AGTTTTCACCGATGTTGTTGG |
| Zm00001d 026018-R | TTCCTTGTTGTGTTTCTGCCC |
| Zm00001d 002540-F | TGCGGGGTTGAAGGAAATGT |
| Zm00001d 002540-R | TGCGGGGTTGAAGGAAATGT |
| Zm00001d 043025-F | ATGAAAAGGCTGCCACGAGA |
| Zm00001d 043025-R | GCTCCAGTATCTTCCCCTC |
| Zm00001d 022017-F | TGACCTCTGTACGCCTCTT |
| Zm00001d 022017-R | GGCTCTCCATTTCCTCTTTGTC |
| Zm00001d 043350-F | GCGGTTCTTGAAGTGGTTGCTC |
| Zm00001d 043350-R | CGAGGACAAGACCAGGAACT |
| Zm00001d 014250-F | GATGGGGTGGGAGGAGATGA |
| Zm00001d 014250-R | CCAGAAGAGGAAGGAGAGGAT |
| UBQ9-F | TACAGTTCTACAAGGTGGACGAC |
| UBQ9-R | GCAGTAGTGGCGGTCGAAGT |
| FVEG_06768-F | GTTATTTCCGCCGCCCTTTCA |
| FVEG_06768-R | GCCACCACCAGCCATACTCGT |
| FVEG_06181-F | GAGTCGCACAAAGAACAAGGA |
| FVEG_06181-R | TACAGAAGGATAGGGAAAGCA |
| FVEG_04041-F | CTTCACTCCCGACGACCCCAA |
| FVEG_04041-R | TATGCCCACATCAGAAACCAG |
| FVEG_06896-F | CTGACGCTTCTACTCCTCTCG |
| FVEG_06896-R | GGGTCTCAATGCCCTCAACTC |
| FVEG_07280-F | CCTGTTCTGATTGGTGGTAAG |
| FVEG_07280-R | CCATCGTTTTTGAGTGGTTCC |
| FVEG_09248-F | CTCAGCGGAACCACCACCCCT |
| FVEG_09248-R | ACCTCGGACAGCAACAGCACG |
| Actin-F | TGCTCCTGAGGCTCTCTTCCA |
| Actin-R | AAGCAAGAATAGAACCACCGA |
表2
转录组测序数据统计"
| 文库 Library | 原始测序数据的 总条目数 Raw reads | 质控后测序数据的 总条目数 Clean reads | 比对到玉米基因组的 百分比 Total mapped rate (maize) (%) | 比对到拟轮枝镰孢 基因组的百分比 Total mapped rate (Fv) (%) | 准确率在99.9%以上的 碱基所占百分比 Q30 (%) |
|---|---|---|---|---|---|
| Mock_1 | 51587734 | 51110882 | 94.63 | - | 95.41 |
| Mock_2 | 50799662 | 50311356 | 95.17 | - | 95.44 |
| Mock_3 | 44878654 | 44387490 | 94.91 | - | 95.12 |
| Fv_1 | 46302536 | 45922668 | - | 95.19 | 95.29 |
| Fv_2 | 50383740 | 49966126 | - | 95.28 | 95.45 |
| Fv_3 | 41064824 | 40694544 | - | 95.28 | 95.42 |
| Fv4_1 | 102436676 | 101474514 | 95.21 | 0.05 | 95.06 |
| Fv4_2 | 96101768 | 95247160 | 95.13 | 0.05 | 95.26 |
| Fv4_3 | 84949414 | 83492592 | 94.76 | 0.04 | 93.59 |
| Fv12_1 | 103162526 | 102019788 | 95.00 | 0.07 | 95.27 |
| Fv12_2 | 107524620 | 106427588 | 95.46 | 0.05 | 95.37 |
| Fv12_3 | 107586942 | 106395294 | 95.25 | 0.06 | 95.20 |
| Fv72_1 | 108575700 | 107478816 | 93.31 | 2.27 | 95.27 |
| Fv72_2 | 104267510 | 103103634 | 91.63 | 2.27 | 95.43 |
| Fv72_3 | 82027324 | 81228748 | 92.25 | 3.21 | 95.30 |
| -:未比对 Not blast | |||||

图1
拟轮枝镰孢中样本间基因表达和差异表达基因的维恩图 A:样本间基因表达的维恩图Venn of gene expression among samples;B:接种4、12和72 h拟轮枝镰孢中差异基因的维恩图Venn of DEGs in F. verticillioides after inoculation at 4, 12 and 72 h;C:接种4、12和72 h后拟轮枝镰孢中上调表达差异基因的维恩图Venn of up-regulated DEGs in F. verticillioides after inoculation at 4, 12 and 72 h;D:接种4、12 和72 h后拟轮枝镰孢中下调表达差异基因的维恩图Venn of down-regulated DEGs in F. verticillioides after inoculation at 4, 12 and 72 h"
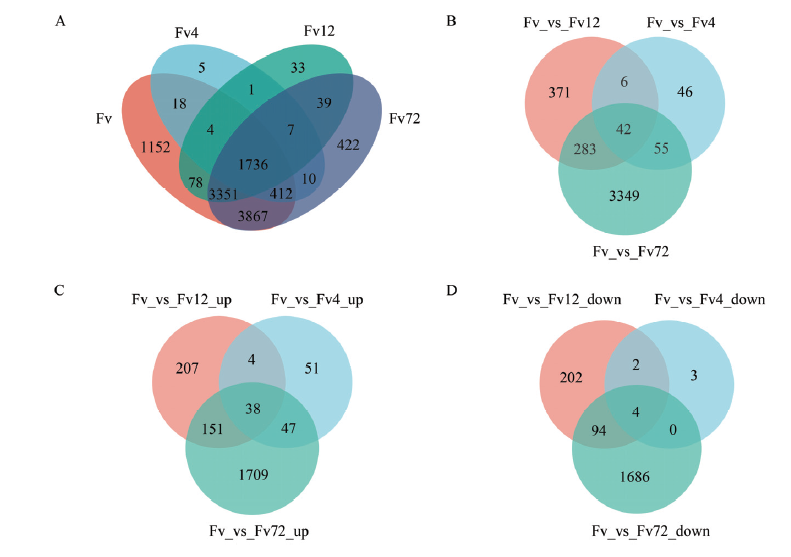
图2
玉米中样本间基因表达和差异表达基因的维恩图 A:样本间基因表达的维恩图Venn of gene expression among samples;B:接种4、12和72 h玉米中差异基因的维恩图Venn of DEGs in maize after inoculation with F. verticillioides at 4, 12 and 72 h;C:接种4、12和72 h后玉米中上调表达差异基因的维恩图Venn of up-regulated DEGs in maize after inoculation with F. verticillioides at 4, 12 and 72 h;D:接种4、12和72 h后玉米中下调表达差异基因的维恩图Venn of down-regulated DEGs in maize after inoculation with F. verticillioides at 4, 12 and 72 h"
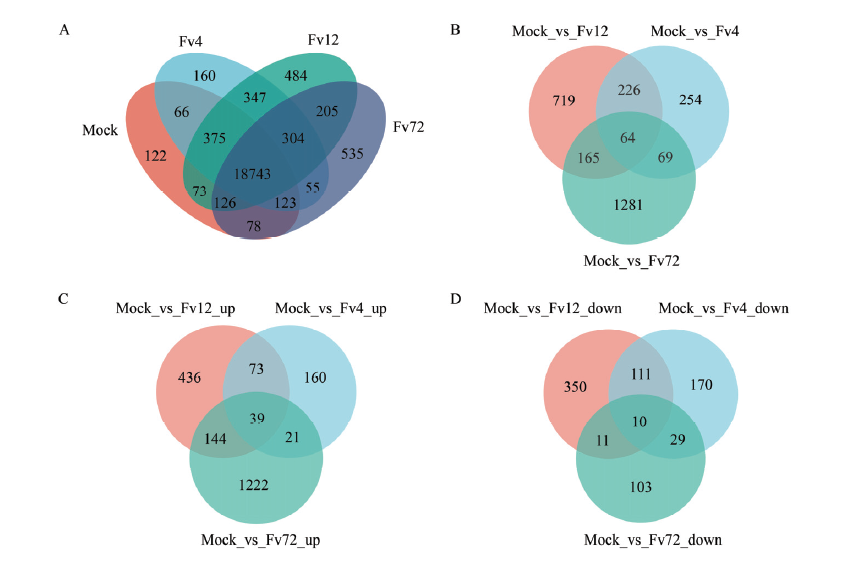
表3
拟轮枝镰孢侵染玉米不同时间点差异表达基因的 GO富集分析"
| 侵染时间Infection time | GO ID | GO功能描述 GO description | 功能类别 Term type | 基因数量 Gene number | P value |
|---|---|---|---|---|---|
| 4 h | GO:0032774 | RNA生物合成过程RNA biosynthetic process | BP | 9 | 0.042121302 |
| GO:0034248 | 细胞酰胺代谢过程的调控Regulation of cellular amide metabolic process | BP | 2 | 0.017806275 | |
| GO:0006417 | 调控翻译Regulation of translation | BP | 2 | 0.016413092 | |
| GO:0005198 | 结构分子活性Structural molecule activity | MF | 5 | 0.023663705 | |
| GO:0004553 | 水解酶活性,水解 O-糖基化合物 Hydrolase activity, hydrolyzing O-glycosyl compounds | MF | 6 | 0.030782616 | |
| GO:0016798 | 水解酶活性,作用于糖基键Hydrolase activity, acting on glycosyl bonds | MF | 6 | 0.042898824 | |
| GO:0005618 | 细胞壁Cell wall | CC | 4 | 1.60E-06 | |
| GO:0009277 | 真菌类型的细胞壁Fungal-type cell wall | CC | 3 | 1.20E-05 | |
| GO:0000786 | 核小体Nucleosome | CC | 2 | 0.003814409 | |
| 12 h | GO:0043043 | 肽生物合成过程Peptide biosynthetic process | BP | 75 | 4.61E-07 |
| GO:0006518 | 肽代谢过程Peptide metabolic process | BP | 75 | 5.47E-07 | |
| GO:0019538 | 蛋白质代谢过程Protein metabolic process | BP | 105 | 2.42E-06 | |
| GO:0005975 | 碳水化合物代谢过程Carbohydrate metabolic process | BP | 44 | 2.92E-06 | |
| GO:0044238 | 初级代谢过程Primary metabolic process | BP | 205 | 4.42E-06 | |
| GO:0008150 | 生物过程Biological process | BP | 359 | 4.94E-06 | |
| GO:0008152 | 代谢过程Metabolic process | BP | 263 | 5.73E-06 | |
| GO:0005198 | 结构分子活性Structural molecule activity | MF | 77 | 7.92E-07 | |
| GO:0003735 | 核糖体的结构成分Structural constituent of ribosome | MF | 71 | 9.56E-07 | |
| GO:0016798 | 水解酶活性Hydrolase activity | MF | 27 | 0.002644 | |
| GO:0003723 | RNA结合RNA binding | MF | 35 | 0.000165 | |
| GO:0032991 | 蛋白质复合物Protein-containing complex | CC | 108 | 0.000515 | |
| GO:0044464 | 细胞部分Cell part | CC | 183 | 5.33E-05 | |
| 72 h | GO:0005975 | 碳水化合物代谢过程Carbohydrate metabolic process | BP | 177 | 2.74E-10 |
| GO:0044347 | 细胞壁多糖分解代谢过程Cell wall polysaccharide catabolic process | BP | 13 | 0.000117 | |
| GO:0055085 | 跨膜运输Transmembrane transport | BP | 366 | 4.16E-10 | |
| GO:0003824 | 催化活性Catalytic activity | MF | 1430 | 4.38E-05 | |
| GO:0022857 | 跨膜转运蛋白活性Transmembrane transporter activity | MF | 239 | 2.10E-08 | |
| GO:0016491 | 氧化还原酶活性Oxidoreductase activity | MF | 466 | 5.60E-08 | |
| GO:0005215 | 转运蛋白活性Transporter activity | MF | 243 | 7.30E-08 | |
| GO:0016021 | 膜的组成部分Integral component of membrane | CC | 1080 | 1.25E-07 | |
| GO:0044425 | 膜部分Membrane part | CC | 1090 | 4.33E-08 |
图3
拟轮枝镰孢中差异表达基因KEGG代谢通路富集分析"

表4
玉米响应拟轮枝镰孢侵染不同时间点差异表达基因的GO富集分类"
| 侵染时间 Infection time | GO ID | GO功能描述 GO description | 功能类别 Term type | 基因数量 Gene number | P value |
|---|---|---|---|---|---|
| 4 h | GO:0042542 | 对过氧化氢的反应Response to hydrogen peroxide | BP | 15 | 3.90E-11 |
| GO:0000302 | 对活性氧的反应Response to reactive oxygen species | BP | 16 | 5.02E-11 | |
| GO:0009628 | 对非生物刺激的反应Response to abiotic stimulus | BP | 52 | 5.25E-11 | |
| GO:0004497 | 单加氧酶活性Monooxygenase activity | MF | 25 | 3.31E-08 | |
| GO:0016491 | 氧化还原酶活性Oxidoreductase activity | MF | 73 | 1.69E-06 | |
| GO:0004568 | 几丁质酶活性Chitinase activity | MF | 6 | 5.52E-05 | |
| GO:0022857 | 跨膜转运蛋白活性Transmembrane transporter activity | MF | 48 | 0.00040527 | |
| 12 h | GO:0042542 | 对过氧化氢的反应Response to hydrogen peroxide | BP | 15 | 5.02E-07 |
| GO:0009628 | 对非生物刺激的反应Response to abiotic stimulus | BP | 85 | 2.20E-06 | |
| GO:0009808 | 木质素代谢过程Lignin metabolic process | BP | 6 | 0.005347 | |
| GO:0004568 | 几丁质酶活性Chitinase activity | MF | 13 | 0.002782 | |
| 72 h | GO:0009699 | 苯丙素生物合成过程Phenylpropanoid biosynthetic process | BP | 20 | 2.29E-07 |
| GO:0009809 | 木质素生物合成过程Lignin biosynthetic process | BP | 13 | 2.60E-07 | |
| GO:0009620 | 对真菌的反应Response to fungus | BP | 22 | 3.56E-07 | |
| GO:0072593 | 活性氧代谢过程Reactive oxygen species metabolic process | BP | 29 | 6.85E-07 | |
| GO:0009694 | 茉莉酸代谢过程Jasmonic acid metabolic process | BP | 9 | 1.55E-05 | |
| GO:0009696 | 水杨酸代谢过程Salicylic acid metabolic process | BP | 5 | 5.05E-05 | |
| GO:0008610 | 脂质生物合成过程Lipid biosynthetic process | BP | 54 | 0.000134 | |
| GO:0004568 | 几丁质酶活性Chitinase activity | MF | 11 | 1.46E-06 | |
| GO:0045548 | 苯丙氨酸解氨酶活性Phenylalanine ammonia-lyase activity | MF | 7 | 3.87E-06 | |
| GO:0004601 | 过氧化物酶活性Peroxidase activity | MF | 27 | 1.50E-05 |
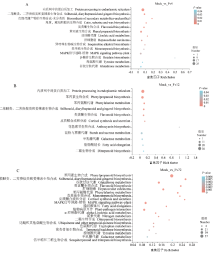
图4
接种拟轮枝镰孢不同时间点后玉米中差异表达基因KEGG代谢通路富集分析"


图5
玉米中植物-抗病通路差异表达基因"

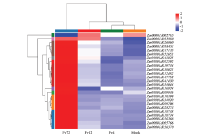
图6
玉米中MAPK信号通路差异表达基因"
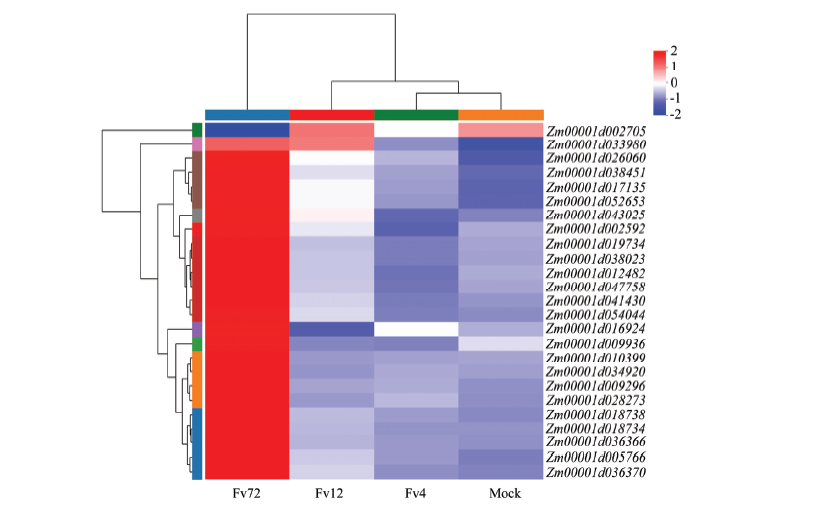
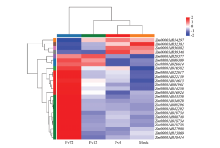
图7
玉米中植物激素信号转导途径差异表达基因"
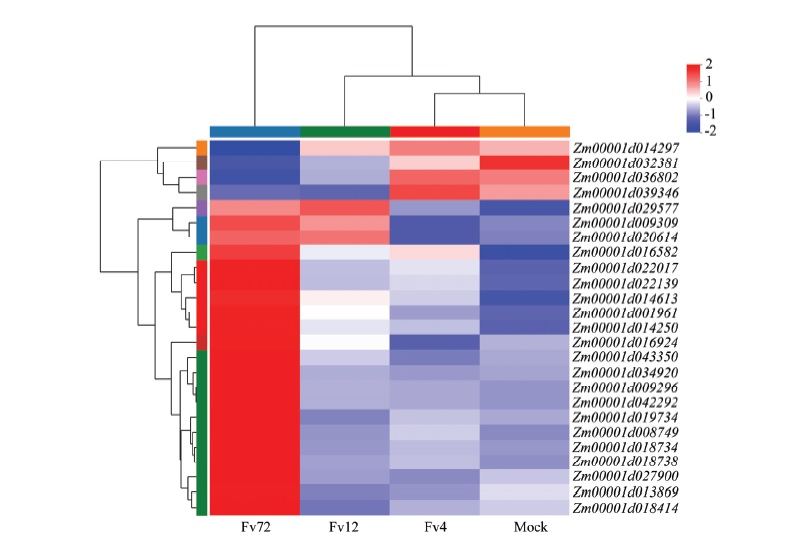

图8
qRT-PCR验证拟轮枝镰孢侵染玉米籽粒72 h后基因表达情况"

| [1] |
doi: 10.3390/toxins8060186 |
| [2] |
doi: 10.1128/AEM.68.5.2101-2105.2002 |
| [3] |
pmid: 16761187 |
| [4] |
doi: 10.1080/02652030601013471 |
| [5] |
doi: 10.1007/BF03192165 pmid: 23605649 |
| [6] |
doi: 10.1016/j.tim.2021.03.019 |
| [7] |
doi: 10.1094/MPMI.2000.13.2.159 |
| [8] |
doi: 10.1016/j.ab.2004.11.004 |
| [9] |
doi: 10.1006/jcrs.2000.0305 |
| [10] |
doi: 10.1023/A:1020847216155 |
| [11] |
doi: 10.1038/nature05286 |
| [12] |
doi: 10.1111/j.1399-3054.2008.01090.x |
| [13] |
doi: 10.1104/pp.106.078717 |
| [14] |
doi: 10.1007/s11103-008-9435-0 pmid: 19083153 |
| [15] |
doi: 10.1016/j.cub.2008.03.060 pmid: 18450451 |
| [16] |
|
| [17] |
doi: 10.1111/mpp.v21.5 |
| [18] |
刘俊. 拟轮枝镰孢和层出镰孢侵染玉米果穗的途径及镰孢菌病害防治[D]. 保定: 河北农业大学, 2017.
|
|
|
|
| [19] |
|
| [20] |
doi: 10.1186/1471-2164-15-710 |
| [21] |
doi: 10.1111/mpp.2015.16.issue-7 |
| [22] |
唐科志, 周常勇. 红橘响应褐斑病菌侵染的转录组学分析. 中国农业科学, 2020, 53(22): 4584-4600.
doi: 10.3864/j.issn.0578-1752.2020.22.006 |
|
|
|
| [23] |
滕彩玲, 钟晰, 吴昊娣, 胡燕, 周常勇, 王雪峰. 马蜂柑响应黄龙病菌不同侵染时期的生物学和转录组学分析. 中国农业科学, 2020, 53(7): 1368-1380.
doi: 10.3864/j.issn.0578-1752.2020.07.007 |
|
|
|
| [24] |
史毅, 牛奎举, 马晖玲. 匍匐翦股颖接种立枯丝核菌后基因表达变化的转录组学分析. 中国农业科学, 2017, 50(17): 3323-3336.
doi: 10.3864/j.issn.0578-1752.2017.17.007 |
|
doi: 10.3864/j.issn.0578-1752.2017.17.007 |
|
| [25] |
doi: 10.1128/AEM.65.8.3668-3673.1999 |
| [26] |
doi: 10.1099/mic.0.27880-0 |
| [27] |
晏石娟, 黄文洁, 刘春明. 脂肪酸及其氧合物对曲霉属真菌菌丝生长、产孢和黄曲霉毒素合成的影响. 微生物学报, 2017, 57(1): 24-32.
|
|
|
|
| [28] |
doi: 10.1016/j.molp.2014.12.022 pmid: 25744358 |
| [29] |
doi: S1360-1385(16)30112-1 pmid: 27666517 |
| [30] |
doi: 10.1016/j.pbi.2010.04.006 pmid: 20471306 |
| [31] |
doi: 10.1038/s41467-019-08726-9 |
| [32] |
doi: 10.1111/jipb.v63.1 |
| [33] |
doi: 10.3389/fpls.2017.01774 |
| [34] |
doi: 10.1111/mpp.2019.20.issue-3 |
| [35] |
doi: 10.1146/annurev-phyto-082712-102314 pmid: 23663002 |
| [36] |
|
| [37] |
doi: 10.3389/fpls.2020.00277 |
| [38] |
doi: 10.1111/tpj.2006.48.issue-4 |
| [39] |
doi: 10.1104/pp.111.192641 |
| [1] | 李懿璞, 童丽秀, 蔺雅楠, 苏治军, 包海柱, 王富贵, 刘剑, 屈佳伟, 胡树平, 孙继颖, 王志刚, 于晓芳, 徐明良, 高聚林. 玉米ZmCCT10耐低氮功能研究[J]. 中国农业科学, 2023, 56(6): 1035-1044. |
| [2] | 汪月宁, 代红军, 贺琰, 魏强, 郭学良, 刘妍, 殷梦婷, 王振平. 基于转录组分析油菜素内酯对高温胁迫下酿酒葡萄花色苷合成及果实品质的调控机制[J]. 中国农业科学, 2023, 56(6): 1139-1153. |
| [3] | 王建锋, 成嘉欣, 舒伟学, 张艳茹, 王晓杰, 康振生, 汤春蕾. 小麦条锈菌效应蛋白Hasp83在条锈菌致病性中的功能分析[J]. 中国农业科学, 2023, 56(5): 866-878. |
| [4] | 彭佳伟, 张叶, 寇单单, 杨丽, 刘晓飞, 张学英, 陈海江, 田义. ‘仓方早生’桃及其早熟芽变不同发育时期果实的转录组分析[J]. 中国农业科学, 2023, 56(5): 964-980. |
| [5] | 邹婷, 刘丽莉, 向建华, 周定港, 吴金锋, 李莓, 李宝, 张大为, 严明理. 芸薹属植物MYBL2基因的克隆及其在A、B、C基因组中的PCR鉴别[J]. 中国农业科学, 2023, 56(3): 416-429. |
| [6] | 张克坤,陈可钦,李婉平,乔浩蓉,张俊霞,刘凤之,房玉林,王海波. 灌水量对限根栽培‘阳光玫瑰’葡萄果实发育与香气物质积累的影响[J]. 中国农业科学, 2023, 56(1): 129-143. |
| [7] | 古丽旦,刘洋,李方向,成卫宁. 小麦吸浆虫小热激蛋白基因Hsp21.9的克隆及在滞育过程与温度胁迫下的表达特性[J]. 中国农业科学, 2023, 56(1): 79-89. |
| [8] | 束婧婷,单艳菊,姬改革,章明,屠云洁,刘一帆,巨晓军,盛中伟,唐燕飞,李华,邹剑敏. 广西麻鸡m6A甲基转移酶基因表达与肌纤维类型及成肌分化的关系[J]. 中国农业科学, 2022, 55(3): 589-601. |
| [9] | 由玉婉,张雨,孙嘉毅,张蔚. ‘月月粉’月季NAC家族全基因组鉴定及皮刺发育相关成员的筛选[J]. 中国农业科学, 2022, 55(24): 4895-4911. |
| [10] | 郭绍雷,许建兰,王晓俊,宿子文,张斌斌,马瑞娟,俞明亮. 桃XTH家族基因鉴定及其在桃果实贮藏过程中的表达特性[J]. 中国农业科学, 2022, 55(23): 4702-4716. |
| [11] | 郝艳,李晓颍,叶茂,刘亚婷,王天宇,王海静,张立彬,肖啸,武军凯. ‘21世纪’桃与‘久脆’桃及其杂交后代果实挥发性成分特征分析[J]. 中国农业科学, 2022, 55(22): 4487-4499. |
| [12] | 尤佳玲,李有梅,孙孟豪,谢兆森. ‘黑比诺’葡萄不同叶龄叶片叶绿体内淀粉积累及其相关基因表达差异分析[J]. 中国农业科学, 2022, 55(21): 4265-4278. |
| [13] | 孙保娟,汪瑞,孙光闻,王益奎,李涛,宫超,衡周,游倩,李植良. 转录组及代谢组联合解析茄子果色上位遗传效应[J]. 中国农业科学, 2022, 55(20): 3997-4010. |
| [14] | 刘鑫,张亚红,袁苗,党仕卓,周娟. ‘红地球’葡萄花芽分化过程中的转录组分析[J]. 中国农业科学, 2022, 55(20): 4020-4035. |
| [15] | 康忱,赵雪芳,李亚栋,田哲娟,王鹏,吴志明. 黄瓜CC-NBS-LRR家族基因鉴定及在霜霉病和白粉病胁迫下的表达分析[J]. 中国农业科学, 2022, 55(19): 3751-3766. |
|
||