| [1] |
KLEIN A M, VAISSIÈRE B E, CANE J H, STEFFAN-DEWENTER I, CUNNINGHAM S A, KREMEN C, TSCHARNTKE T . Importance of pollinators in changing landscapes for world crops. Proceedings of the Royal Society B-Biological Sciences, 2007,274(1608):303-313.
|
| [2] |
POTTS S G, BIESMEIJER J C, KREMEN C, NEUMANN P, SCHWEIGER O, KUNIN W E . Global pollinator declines: trends, impacts and drivers. Trends in Ecology and Evolution, 2010,25(6):345-353.
|
| [3] |
曾志将, 颜伟玉, 饶波, 谢宪斌 . 蜜蜂性比研究进展. 上海交通大学学报(农业科学版), 2003,21(2):164-167.
|
|
ZENG Z J, YAN W Y, RAO B, XIE X B . Advances on the honeybee sex ratio. Journal of Shanghai Jiaotong University (Agricultural Science), 2003,21(2):164-167. (in Chinese)
|
| [4] |
TRAVER B E, FELL R D . Low natural levels of Nosema ceranae in Apis mellifera queens. Journal of Invertebrate Pathology, 2012,110(3):408-410.
|
| [5] |
TRAVER B E, FELL R D . Nosema ceranae in drone honey bees(Apis mellifera). Journal of Invertebrate Pathology, 2011,107(3):234-236.
|
| [6] |
GARCIA-PALENCIA P, MARTIN-HERNANDEZ R, GONZALEZ- PORTO A V, MARIN P, MEANA A, HIGES M . Natural infection by Nosema ceranae causes similar lesions as in experimentally infected caged-worker honey bees(Apis mellifera). Journal of Apicultural Research, 2010,49(3):278-283.
|
| [7] |
MAYACK C, NAUG D . Energetic stress in the honeybee Apis mellifera from Nosema ceranae infection. Journal of Invertebrate Pathology, 2009,100(3):185-188.
|
| [8] |
MARTIN-HERNANDEZ R, BOTIAS C, BARRIOS L, MARTINEZ- SALVADOR A, MEANA A, MAYACK C, HIGES M . Comparison of the energetic stress associated with experimental Nosema ceranae and Nosema apis infection of honeybees(Apis mellifera). Parasitology Research, 2011,109(3):605-612.
|
| [9] |
LECOCQ A, JENSEN A B, KRYGER P, NIEH J C . Parasite infection accelerates age polyethism in young honey bees. Scientific Reports, 2016,6:22042.
|
| [10] |
SHUTLER D, HEAD K, BURGHER-MACLELLAN K L, COLWELL M J, LEVITT A L, OSTIGUY N, WILLIAMS G R . Honey bee Apis mellifera parasites in the absence of Nosema ceranae fungi and Varroa destructor mites. PLoS ONE, 2014,9(6):e98599.
|
| [11] |
WU J Y, SMART M D, ANELLI C M, SHEPPARD W S . Honey bees (Apis mellifera) reared in brood combs containing high levels of pesticide residues exhibit increased susceptibility to Nosema(Microsporidia) infection. Journal of Invertebrate Pathology, 2012,109(3):326-329.
|
| [12] |
COX-FOSTER D L, CONLAN S, HOLMES E C, PALACIOS G, EVANS J D, MORAN N A, QUAN P L, BRIESE T, HORNIG M, GEISER D M, MARTINSON V, VANENGELSDORP D, KALKSTEIN A L, DRYSDALE A, HUI J, ZHAI J, CUI L, HUTCHISON S K, SIMONS J F, EGHOLM M, PETTIS J S, LIPKIN W I . A metagenomic survey of microbes in honey bee colony collapse disorder. Science, 2007,318(5848):283-287.
|
| [13] |
CHEJANOVSKY N, OPHIR R, SCHWAGER M S, SLABEZKI Y, GROSSMAN S, COX-FOSTER D . Characterization of viral siRNA populations in honey bee colony collapse disorder. Virology, 2014,454/455:176-183.
|
| [14] |
KRIBS-ZALETA C M, MITCHELL C . Modeling colony collapse disorder in honeybees as a contagion. Mathematical Biosciences and Engineering, 2014,11(6):1275-1294.
|
| [15] |
MAYACK C, NATSOPOULOU M E, MCMAHON D P . Nosema ceranae alters a highly conserved hormonal stress pathway in honeybees. Insect Molecular Biology, 2015,24(6):662-670.
|
| [16] |
NATSOPOULOU M E, DOUBLET V, PAXTON R J . European isolates of the microsporidia Nosema apis and Nosema ceranae have similar virulence in laboratory tests on European worker honey bees. Apidologie, 2016,47(1):57-65.
|
| [17] |
BRAVO J, CARBONELL V, SEPÚLVEDA B, DELPORTE C, VALDOVINOS C E, MARTÍN-HERNÁNDEZ R, HIGES M . Antifungal activity of the essential oil obtained from Cryptocarya alba against infection in honey bees by Nosema ceranae. Journal of Invertebrate Pathology, 2017,149:141-147.
|
| [18] |
CORNMAN R S, CHEN Y P, SCHATZ M C, STREET C, ZHAO Y, DESANY B, EGHOLM M, HUTCHISON S, PETTIS J S, LIPKIN W I, EVANS J D . Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathogens, 2009,5(6):e1000466.
|
| [19] |
BADAOUI B, FOUGEROUX A, PETIT F, ANSELMO A, GORNI C, CUCURACHI M, CERSINI A, GRANATO A, CARDETI G, FORMATO G, MUTINELLI F, GIUFFRA E, WILLIAMS J L, BOTTI S . RNA-sequence analysis of gene expression from honeybees (Apis mellifera) infected with Nosema ceranae. PLoS ONE, 2017,12(3):e0173438.
|
| [20] |
AUFAUVRE J, MISME-AUCOUTURIER B, VIQUES B, TEXIER C, DELBAC F, BLOT N . Transcriptome analyses of the honeybee response to Nosema ceranae and insecticides. PLoS ONE, 2014,9(3):e91686.
|
| [21] |
郭睿, 刀晨, 熊翠玲, 郑燕珍, 付中民, 耿四海, 陈大福 . 意大利蜜蜂工蜂中肠响应Nosema ceranae胁迫的高表达基因分析. 环境昆虫学报, 2018,40(5):1106-1112.
|
|
GUO R, DAO C, XIONG C L, ZHENG Y Z, FU Z M, GENG S H, CHEN D F . Analysis of the highly expressed genes in the midgut of Apis mellifera ligustica worker under the stress of Nosema ceranae. Journal of Environmental Entomology, 2018,40(5):1106-1112. (in Chinese)
|
| [22] |
郭睿, 耿四海, 熊翠玲, 郑燕珍, 付中民, 王海朋, 杜宇, 童新宇, 赵红霞, 陈大福 . 意大利蜜蜂工蜂中肠发育过程中长链非编码RNA的差异表达分析. 中国农业科学, 2018,51(18):3600-3613.
doi: 10.3864/j.issn.0578-1752.2018.18.016
|
|
GUO R, GENG S H, XIONG C L, ZHENG Y Z, FU Z M, WANG H P, DU Y, TONG X Y, ZHAO H X, CHEN D F . Differential expression analysis of long non-coding RNAs during the developmental process of Apis mellifera ligustica worker’s midgut. Scientia Agricultura Sinica, 2018,51(18):3600-3613. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2018.18.016
|
| [23] |
郭睿, 陈华枝, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 杜宇, 王海朋, 耿四海, 周丁丁, 刘思亚, 陈大福 . 意大利蜜蜂工蜂中肠发育过程中的差异表达环状RNA及其调控网络分析. 中国农业科学, 2018,51(23):4575-4590.
doi: 10.3864/j.issn.0578-1752.2018.23.015
|
|
GUO R, CHEN H Z, XIONG C L, ZHENG Y Z, FU Z M, XU G J, DU Y, WANG H P, GENG S H, ZHOU D D, LIU S Y, CHEN D F . Analysis of differentially expressed circular RNAs and their regulation networks during the developmental process of Apis mellifera ligustica worker’s midgut. Scientia Agricultura Sinica, 2018,51(23):4575-4590. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2018.23.015
|
| [24] |
陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 张曌楠, 黄枳腱, 张璐, 王鸿权, 解彦玲, 童新宇 . 中华蜜蜂幼虫肠道响应球囊菌早期胁迫的转录组学. 中国农业科学, 2017,50(13):2614-2623.
doi: 10.3864/j.issn.0578-1752.2017.13.019
|
|
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, ZHANG Z N, HUANG Z J, ZHANG L, WANG H Q, XIE Y L, TONG X Y . Transcriptome of Apis cerana cerana larval gut under the stress of Ascosphaera apis. Scientia Agricultura Sinica, 2017,50(13):2614-2623. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2017.13.019
|
| [25] |
ARONSTEIN K A, MURRAY K D . Chalkbrood disease in honey bees. Journal of Invertebrate Pathology, 2010,103(Suppl. 1):S20-S29.
|
| [26] |
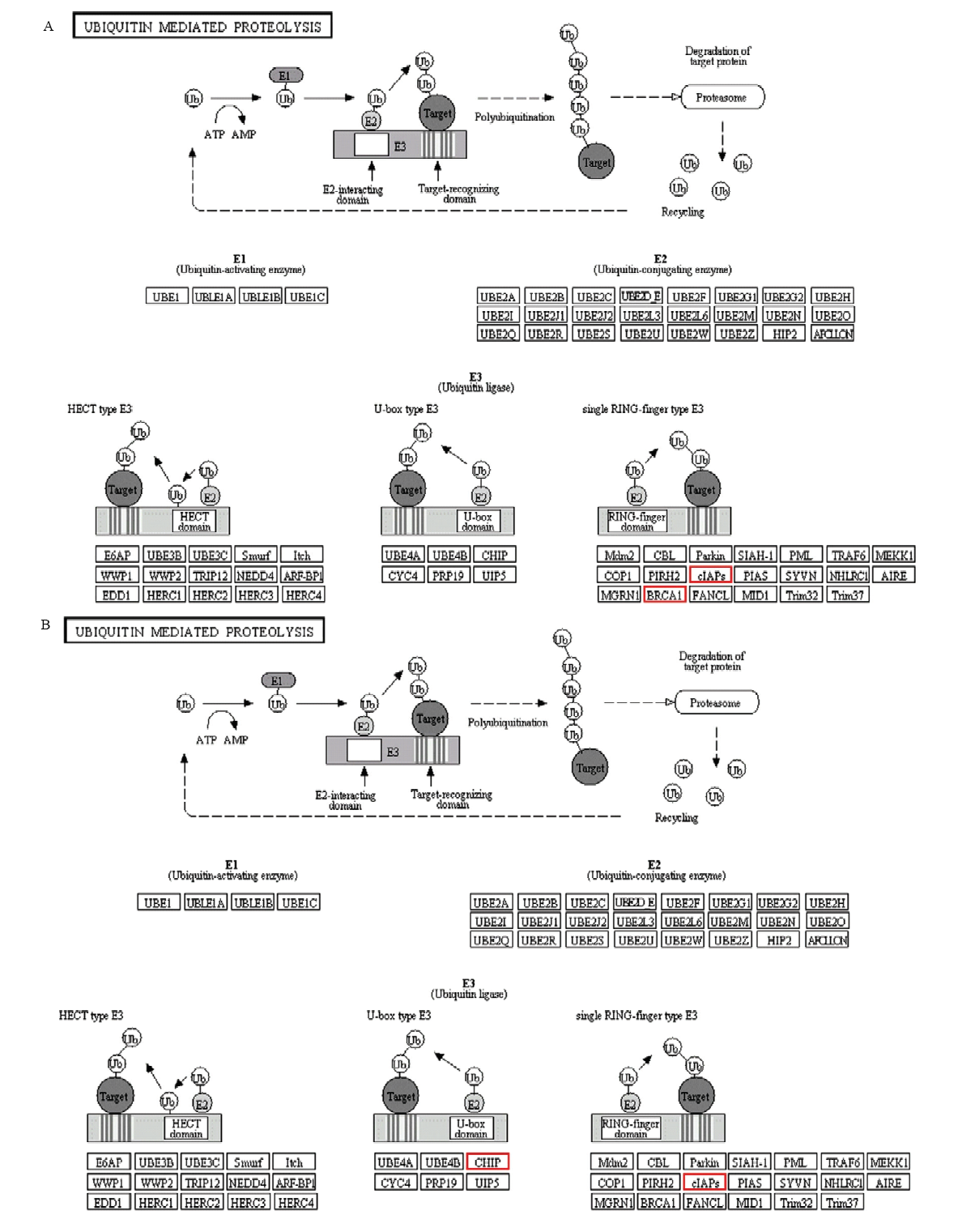
CIECHANOVER A, 葛亮 . 泛素介导的蛋白质降解系统——从基础研究到临床应用. 生命科学, 2010,22(3):212-215.
|
|
CIECHANOVER A, GE L . The ubiquitin proteolytic system—from bench to the bedside. Chinese Bulletin of Life Sciences, 2010,22(3):212-215. (in Chinese)
|
| [27] |
李朝飞, 庞义 . 泛素-蛋白水解酶复合体通路与病毒侵染. 生物工程学报, 2004,20(2):151-156.
|
|
LI C F, PANG Y . Ubiquitin-proteasome pathway and virus infection. Chinese Journal of Biotechnology, 2004,20(2):151-156. (in Chinese)
|
| [28] |
杜宇, 童新宇, 周丁丁, 陈大福, 熊翠玲, 郑燕珍, 徐国钧, 王海朋, 陈华枝, 郭意龙, 隆琦, 郭睿 . 中华蜜蜂幼虫肠道响应球囊菌胁迫的microRNA应答分析. 微生物学报, 2019. DOI: 10.13343/j.cnki.wsxb.20180401.
|
|
DU Y, TONG X Y, ZHOU D D, CHEN D F, XIONG C L, ZHENG Y Z, XU G J, WANG H P, CHEN H Z, GUO Y L, LONG Q, GUO R . MicroRNA responses in the larval gut of Apis cerana cerana to Ascosphaera apis stress. Acta Microbiologica Sinica, 2019. DOI: 10.13343/j.cnki.wsxb.20180401. (in Chinese)
|
| [29] |
陈阳 . 鸭RIG-I在免疫调节中的作用及分子调控机制[D]. 扬州: 扬州大学, 2015.
|
|
CHEN Y . Function and molecular mechanism of duck RIG-I gene in immune regulation[D]. Yangzhou: Yangzhou University, 2015. (in Chinese)
|
| [30] |
徐娇 . miR-964在果蝇Toll免疫响应中的调控作用研究[D]. 南京: 南京师范大学, 2018.
|
|
XU J . Study on the regulation of miR-964 in the immune response of drosophila Toll[D]. Nanjing: Nanjing Normal University, 2018. (in Chinese)
|
| [31] |
SALMENA L, POLISENO L, TAY Y, KATS L, PANDOLFI P P . A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell, 2011,146(3):353-358.
|







 )
)