






中国农业科学 ›› 2021, Vol. 54 ›› Issue (6): 1288-1300.doi: 10.3864/j.issn.0578-1752.2021.06.018
• 畜牧·兽医·资源昆虫 • 上一篇
陈华枝1( ),范元婵1(),蒋海宾1,王杰1,范小雪1,祝智威1,隆琦1,蔡宗兵1,郑燕珍1,付中民1,2,徐国钧1,陈大福1,2,郭睿1,2()
),范元婵1(),蒋海宾1,王杰1,范小雪1,祝智威1,隆琦1,蔡宗兵1,郑燕珍1,付中民1,2,徐国钧1,陈大福1,2,郭睿1,2()
收稿日期:2020-05-06
接受日期:2020-05-28
出版日期:2021-03-16
发布日期:2021-03-25
联系方式:
陈华枝,E-mail:CHZ0720@outlook.com。|范元婵,E-mail:fanyc19980201@126.com。
基金资助:
HuaZhi CHEN1(),YuanChan FAN1(),HaiBin JIANG1,Jie WANG1,XiaoXue FAN1,ZhiWei ZHU1,Qi LONG1,ZongBing CAI1,YanZhen ZHENG1,ZhongMin FU1,2,GuoJun XU1,DaFu CHEN1,2,Rui GUO1,2()
Received:2020-05-06
Accepted:2020-05-28
Published:2021-03-16
Online:2021-03-25
摘要:
【目的】利用已获得的纳米孔全长转录组数据对现有的东方蜜蜂微孢子虫(Nosema ceranae)参考基因组的基因序列和功能注释进行完善。【方法】采用TransDecoder软件预测东方蜜蜂微孢子虫基因的开放阅读框(open reading frame,ORF)及相应的氨基酸。利用gffcompare软件将全长转录本与参考基因组注释的转录本进行比较,对基因组注释基因的非编码区向上游或下游延伸,修正基因的边界。利用MISA软件鉴定长度在500 bp以上的全长转录本的简单重复序列(simple sequence repeat,SSR)位点,包括单核苷酸重复、双核苷酸重复、三核苷酸重复、四核苷酸重复、五核苷酸重复、六核苷酸重复、混合SSR等类型。通过Blast工具将鉴定到的新基因和新转录本比对Nr、KOG、eggNOG、GO和KEGG数据库,从而获得功能注释。【结果】共预测出2 353个完整ORF,其中长度分布在0—100个氨基酸的ORF最多,占总ORF数的72.12%。共对东方蜜蜂微孢子虫的2 340个基因进行了结构优化,其中5′端延长的基因有1 182个,3′端延长的基因有1 158个。共鉴定到1 658个SSR,其中单核苷酸重复、双核苷酸重复、三核苷酸重复、四核苷酸重复的数量分别为1 622、23、7和6个;单核苷酸重复类型的SSR密度最大,达到182.32个/Mb,其次为混合SSR、双核苷酸重复和三核苷酸重复,分别达到6.90、2.78和0.73个/Mb。共鉴定出954个新基因,其中分别有951、333、371、422和321个新基因可注释到Nr、KOG、eggNOG、GO和KEGG数据库。此外,还鉴定出6 164条新转录本,其中分别有6 141、2 808、2 932、3 196和2 585条新转录本可注释到Nr、KOG、eggNOG、GO和KEGG数据库。新基因和新转录本注释数量最多的物种均为东方蜜蜂微孢子虫,其次是蜜蜂微孢子虫(Nosema apis)。【结论】研究结果较好地完善了现有的东方蜜蜂微孢子虫参考基因组已注释基因的序列和功能注释,并补充和注释了大量参考基因组未注释的新基因和新转录本。
陈华枝,范元婵,蒋海宾,王杰,范小雪,祝智威,隆琦,蔡宗兵,郑燕珍,付中民,徐国钧,陈大福,郭睿. 基于纳米孔全长转录组数据完善东方蜜蜂微孢子虫的基因组注释[J]. 中国农业科学, 2021, 54(6): 1288-1300.
HuaZhi CHEN,YuanChan FAN,HaiBin JIANG,Jie WANG,XiaoXue FAN,ZhiWei ZHU,Qi LONG,ZongBing CAI,YanZhen ZHENG,ZhongMin FU,GuoJun XU,DaFu CHEN,Rui GUO. Improvement of Nosema ceranae Genome Annotation Based on Nanopore Full-Length Transcriptome Data[J]. Scientia Agricultura Sinica, 2021, 54(6): 1288-1300.
表1
东方蜜蜂微孢子虫参考基因组中10个基因的结构优化信息概要"
| 基因ID Gene ID | 基因位置 Gene locus | 正负链 Plus and minus strand | 末端 End | 原位置 Original site | 优化后位置 Optimized site |
|---|---|---|---|---|---|
| Gene1175 | NW_020169317.1:49652-50638 | - | 3′ | 50341 | 50638 |
| Gene326 | NW_020169413.1:1478-3041 | - | 3′ | 2683 | 3041 |
| Gene503 | NW_020169431.1:1208-2193 | - | 5′ | 1461 | 1208 |
| Gene503 | NW_020169431.1:1208-2193 | - | 3′ | 1634 | 2193 |
| Gene1591 | NW_020169298.1:96744-98975 | + | 5′ | 96746 | 96744 |
| Gene1591 | NW_020169298.1:96744-98975 | + | 3′ | 98671 | 98975 |
| Gene588 | NW_020169310.1:2436-3030 | + | 3′ | 2741 | 3030 |
| Gene350 | NW_020169307.1:13073-18338 | - | 5′ | 17277 | 13073 |
| Gene36 | NW_020169296.1: 54153-55245 | - | 5′ | 54289 | 54153 |
| Gene2363 | NW_020169300.1:40046-41314 | - | 5′ | 40510 | 40046 |
表2
基于MISA的东方蜜蜂微孢子虫SSR的搜索结果"
| MISA搜索项目 MISA searching item | 数量 Number |
|---|---|
| 搜索基因 Searching gene | 8021 |
| 搜索基因的总序列长度 Total sequence length of searching gene (bp) | 8265494 |
| 鉴定到的SSR位点 Identified SSR loci | 1658 |
| 鉴定到的SSR总序列长度 Total sequence length of identified SSRs (bp) | 1405 |
| 含有1个以上SSR的基因 Genes containing more than one SSR | 212 |
| 混合SSR Mixed SSR | 65 |
| 单核苷酸重复Single nucleotide repeat | 1622 |
| 双核苷酸重复Dinucleotide repeat | 23 |
| 三核苷酸重复Trinucleotide repeat | 7 |
| 四核苷酸重复Tetranucleotide repeat | 6 |
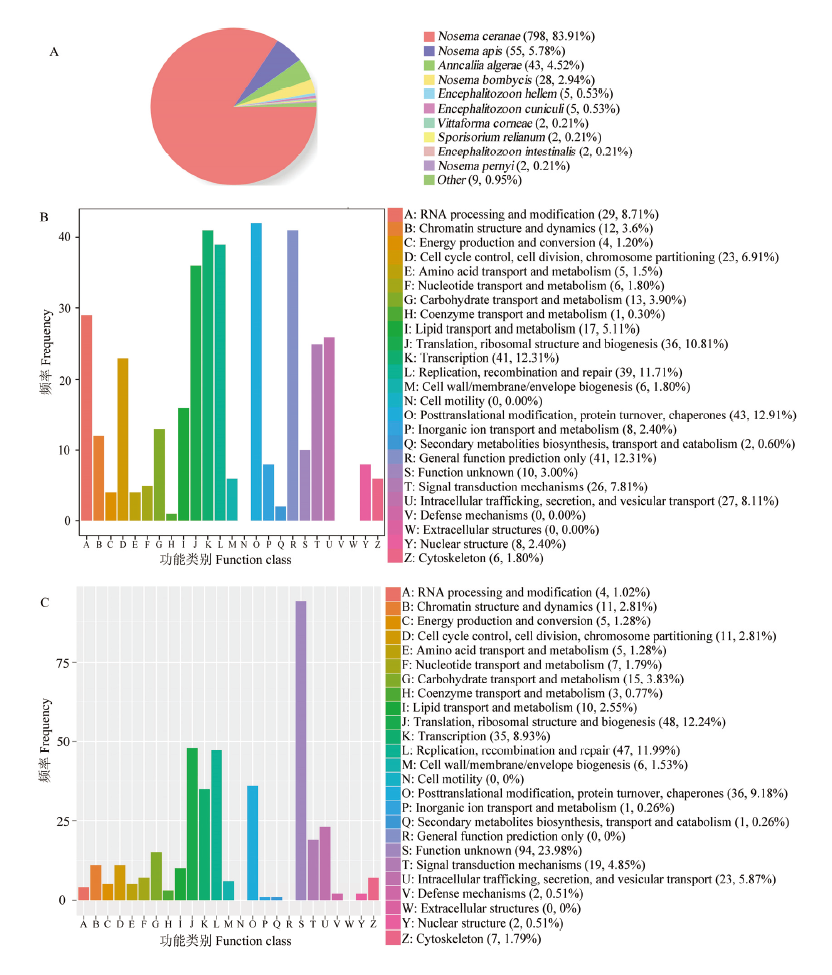
图1
东方蜜蜂微孢子虫新基因的Nr(A)、KOG(B)和eggNOG(C)数据库注释"
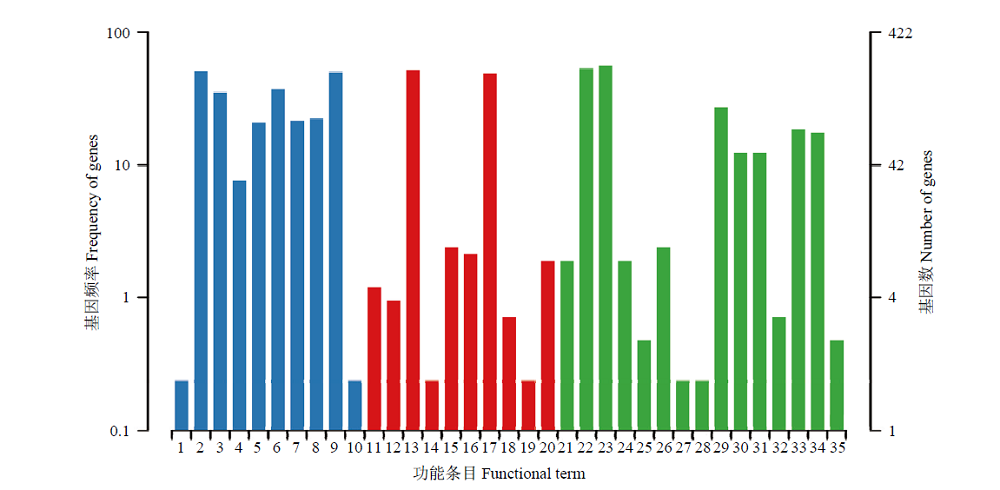
图2
东方蜜蜂微孢子虫新基因的GO数据库注释 1:细胞外区域Extracellular region;2:细胞Cell;3:细胞膜Cell membrane;4:细胞膜内腔Membrane-enclosed lumen;5:高分子复合物Macromolecular complex;6:细胞器Organelle;7:细胞器组件Organelle part;8:细胞膜组件Cell membrane part;9:细胞组件Cell part;10:超分子复合物Supramolecular complex;11:转录因子活性,蛋白质结合Transcription factor activity, protein binding;12:核酸结合转录因子活性Nucleic acid binding transcription factor activity;13:催化活性Catalytic activity;14:信号转导因子活性Signal transducer activity;15:结构分子活性Structural molecule activity;16:转运活性Transporter activity;17:结合Binding;18:电子载体活性Electron carrier activity;19:抗氧化活性Antioxidant activity;20:分子功能调节因子Molecular function regulator;21:繁殖Reproduction;22:代谢进程Metabolic process;23:细胞进程Cellular process;24:生殖进程Reproductive process;25:生物黏附Biological adhesion;26:信号Signaling;27:发育进程Developmental process;28:生长Growth;29:单一组织进程Single-organism process;30:应激反应Response to stimulus;31:定位Localization;32:多组织进程Multi-organism process;33:生物调控Biological regulation;34:细胞成分组织或生物合成Cellular component organization or biogenesis;35:解毒Detoxification"
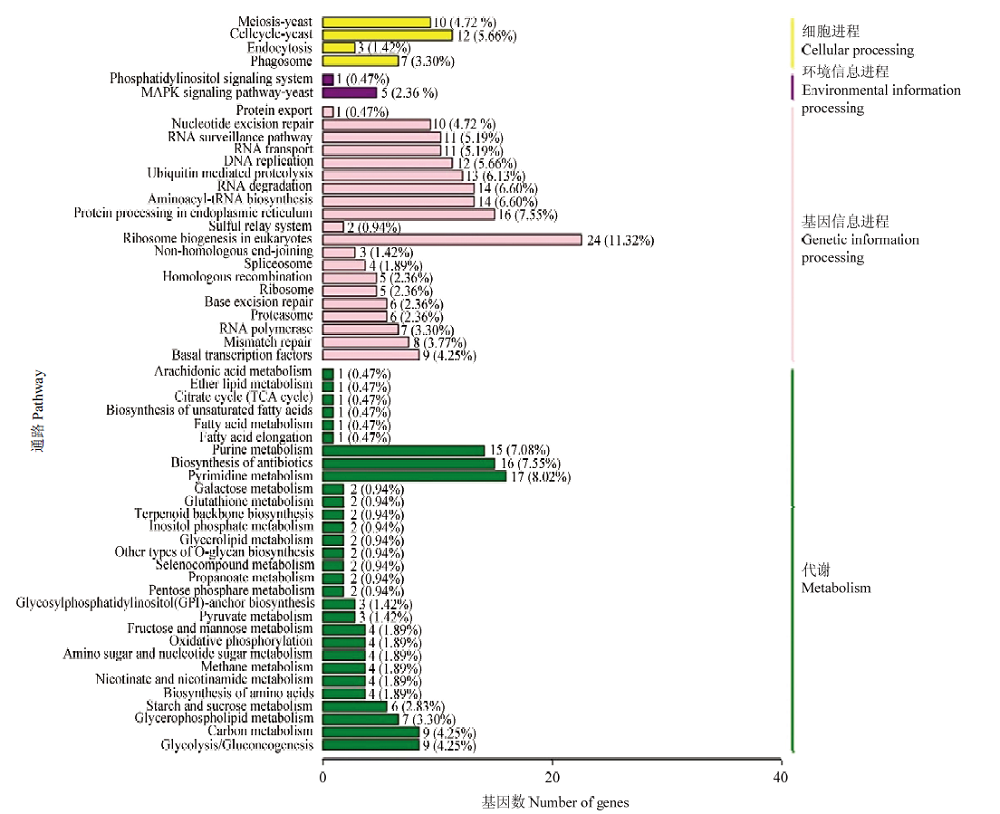
图3
东方蜜蜂微孢子虫新基因的KEGG数据库注释"
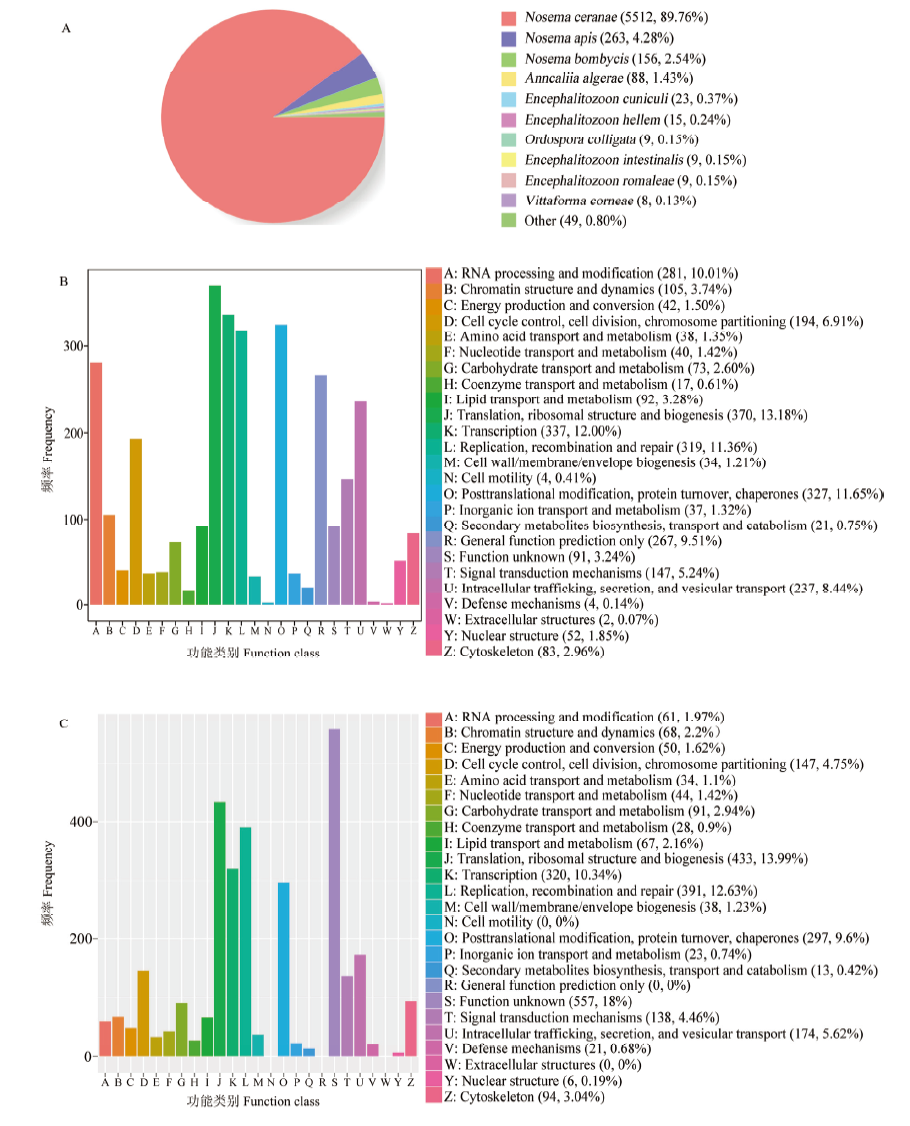
图4
东方蜜蜂微孢子虫新转录本的Nr(A)、KOG(B)和eggNOG(C)数据库注释"
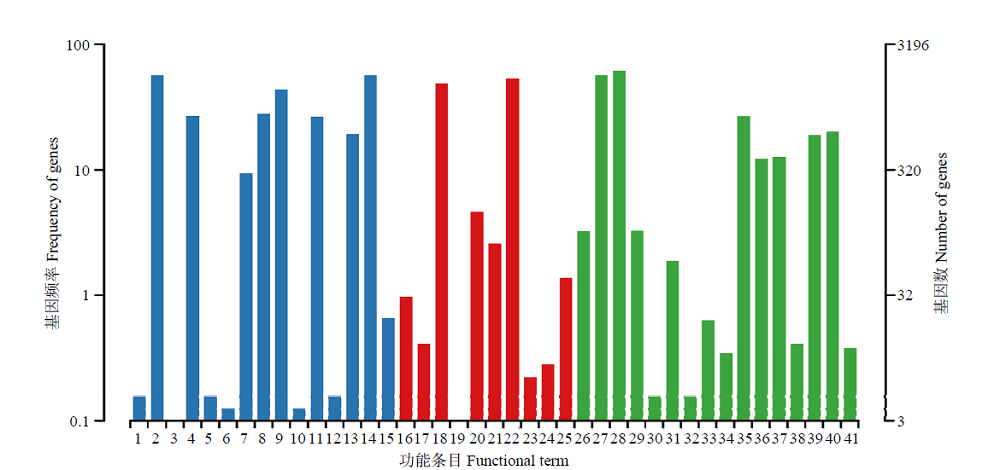
图5
东方蜜蜂微孢子虫新转录本的GO数据库注释 1:细胞外区域Extracellular region;2:细胞Cell;3:拟核Nucleoid;4:细胞膜Cell membrane;5:病毒Virion;6:细胞连接Cell junction;7:细胞膜内腔Membrane-enclosed lumen;8:高分子复合物Macromolecular complex;9:细胞器Organelle;10:胞外区组件Extracellular region part;11:细胞器组件Organelle part;12:病毒组件Virion part;13:细胞膜组件Cell membrane part;14:细胞组件Cell part;15:超分子复合物Supramolecular complex;16:转录因子活性,蛋白质结合Transcription factor activity, protein binding;17:核酸结合转录因子活性Nucleic acid binding transcription factor activity;18:催化活性Catalytic activity;19:信号转导因子活性Signal transducer activity;20:结构分子活性Structural molecule activity;21:转运活性Transporter activity;22:结合Binding;23:电子载体活性Electron carrier activity;24:分子功能调控器Molecular function regulator;25:抗氧化活性Antioxidant activity;26:繁殖Reproduction;27:代谢进程Metabolic process;28:细胞进程Cellular process;29:生殖进程Reproductive process;30:生物黏附Biological adhesion;31:信号Signaling;32:多组织进程Multicellular organismal process;33:发育进程Developmental process;34:生长Growth;35:单一组织进程Single-organism process;36:应激反应Response to stimulus;37:定位Localization;38:多细胞组织进程Multi-organism process;39:生物调控Biological regulation;40:细胞成分组织或生物合成Cellular component organization or biogenesis;41:解毒Detoxification"
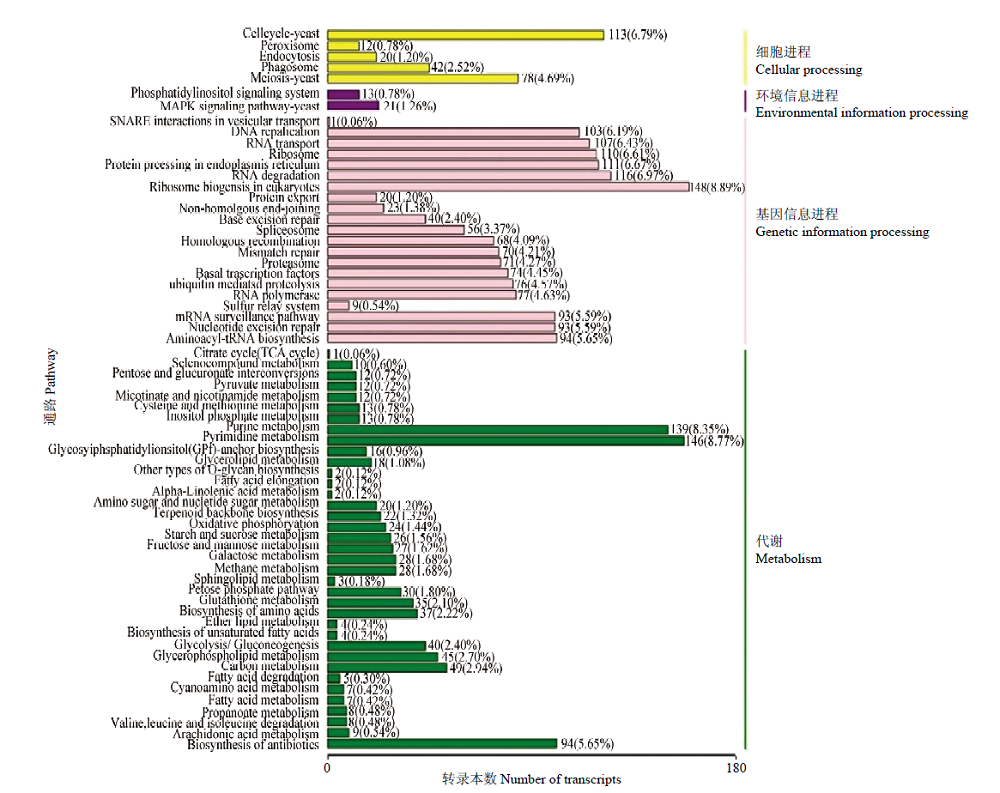
图6
东方蜜蜂微孢子虫新转录本的KEGG数据库注释"
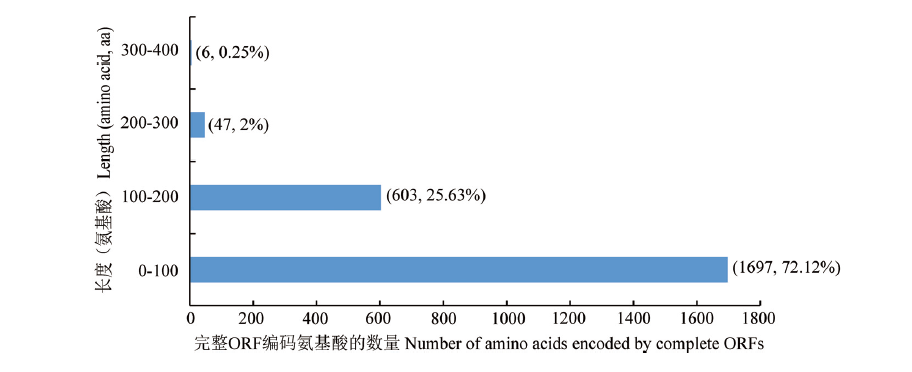
图7
东方蜜蜂微孢子虫的完整ORF编码氨基酸的长度分布"
| [1] |
WHEELER D A, SRINIVASAN M, EGHOLM M, SHEN Y, CHEN L, MCGUIRE A, HE W, CHEN Y J, MAKHIJANI V, ROTH G T, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature, 2008,452(7189):872-876.
doi: 10.1038/nature06884 pmid: 18421352 |
| [2] |
XIA Q, ZHOU Z, LU C, CHENG D, DAI F, LI B, ZHAO P, ZHA X, CHENG T, CHAI C, et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science, 2004,306(5703):1937-1940.
doi: 10.1126/science.1102210 pmid: 15591204 |
| [3] |
The Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature, 2006,443(7114):931-949.
doi: 10.1038/nature05260 pmid: 17073008 |
| [4] |
KOCHER S D, LI C, YANG W, TAN H, YI S V, YANG X, HOEKSTRA H E, ZHANG G, PIERCE N E, YU D W. The draft genome of a socially polymorphic halictid bee, Lasioglossum albipes. Genome Biology, 2013,14(12):R142.
doi: 10.1186/gb-2013-14-12-r142 pmid: 24359881 |
| [5] |
PARK D, JUNG J W, CHOI B S, JAYAKODI M, LEE J, LIM J, YU Y, CHOI Y S, LEE M L, PARK Y, CHOI I Y, YANG T J, EDWARDS O R, NAH G, KWON H W. Uncovering the novel characteristics of Asian honey bee, Apis cerana, by whole genome sequencing. BMC Genomics, 2015,16(1):1.
doi: 10.1186/1471-2164-16-1 |
| [6] |
OLSEN J L, ROUZÉ P, VERHELST B, LIN Y C, BAYER T, COLLEN J, DATTOLO E, DE PAOLI E, DITTAMI S, MAUMUS F, et al. The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature, 2016,530(7590):331-335.
doi: 10.1038/nature16548 pmid: 26814964 |
| [7] |
ZHANG G Q, XU Q, BIAN C, TSAI W C, YEH C M, LIU K W, YOSHIDA K, ZHANG L S, CHANG S B, CHEN F, et al. The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Scientific Reports, 2016,6:19029.
doi: 10.1038/srep19029 pmid: 26754549 |
| [8] |
PELIN A, SELMAN M, 1 ARIS-BROSOU S, FARINELLI L, CORRADI N. Genome analyses suggest the presence of polyploidy and recent human-driven expansions in eight global populations of the honeybee pathogen Nosema ceranae. Environmental Microbiology, 2015,17(11):4443-4458.
doi: 10.1111/1462-2920.12883 pmid: 25914091 |
| [9] |
LIU B, ZHOU Y, LI K, HU X, WANG C, CAO G, XUE R, GONG C. The complete genome of cyprinid herpesvirus 2, a new strain isolated from Allogynogenetic crucian carp. Virus Research, 2018,256:6-10.
doi: 10.1016/j.virusres.2018.07.016 pmid: 30053416 |
| [10] |
RHOADS A, AU K F. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics, 2015,13(5):278-289.
doi: 10.1016/j.gpb.2015.08.002 pmid: 26542840 |
| [11] |
MAGRINI V, GAO X, ROSA B A, MCGRATH S, ZHANG X, HALLSWORTH-PEPIN K, MARTIN J, HAWDON J, WILSON R K, MITREVA M. Improving eukaryotic genome annotation using single molecule mRNA sequencing. BMC Genomics, 2018,19(1):172.
pmid: 29495964 |
| [12] |
TANG Y T, GAO X, ROSA B A, ABUBUCKER S, HALLSWORTH- PEPIN K, MARTIN J, TYAGI R, HEIZER E, ZHANG X, BHONAGIRI-PALSIKAR V, et al. Genome of the human hookworm Necator americanus. Nature Genetics, 2014,46(3):261-269.
doi: 10.1038/ng.2875 |
| [13] |
MITREVA M, JASMER D P, ZARLENGA D S, WANG Z, ABUBUCKER S, MARTIN J, TAYLOR C M, YIN Y, FULTON L, MINX P, et al. The draft genome of the parasitic nematode Trichinella spiralis. Nature Genetics, 2011,43(3):228-235.
doi: 10.1038/ng.769 pmid: 21336279 |
| [14] |
PENDLETON M, SEBRA R, PANG A W C, UMMAT A, FRANZEN O, RAUSCH T, STÜTZ A M, STEDMAN W, ANANTHARAMAN T, HASTIE A, et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nature Methods, 2015,12(8):780-786.
doi: 10.1038/nmeth.3454 pmid: 26121404 |
| [15] |
SHIELDS E J, SHENG L, WEINER A K, GARCIA B A, BONASIO R. High-quality genome assemblies reveal long non-coding RNAs expressed in ant brains. Cell Reports, 2018,23(10):3078-3090.
doi: 10.1016/j.celrep.2018.05.014 pmid: 29874592 |
| [16] |
DACCORD N, CELTON J M, LINSMITH G, BECKER C, CHOISNE N, SCHIJLEN E, VAN DE GEEST H, BIANCO L, MICHELETTI D, VELASCO R, et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nature Genetics, 2017,49(7):1099-1106.
pmid: 28581499 |
| [17] |
DONG L, LIU H, ZHANG J, YANG S, KONG G, CHU S C, CHEN N, WANG D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genomics, 2015,16(1):1039.
doi: 10.1186/s12864-015-2257-y |
| [18] |
CORNMAN R S, CHEN Y P, SCHATZ M C, STREET S, ZHAO Y, DESANY B, EGHOLM M, HUTCHISON S, PETTIS J S, LIPKIN W I, EVANS J D. Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathogens, 2009,5(6):e1000466.
doi: 10.1371/journal.ppat.1000466 pmid: 19503607 |
| [19] | 陈华枝, 杜宇, 范小雪, 祝智威, 蒋海宾, 王杰, 范元婵, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 陈大福, 郭睿. 基于第三代纳米孔测序技术的东方蜜蜂微孢子虫全长转录组构建及注释. 昆虫学报, 2020,63(12):1461-1472. |
| CHEN H Z, DU Y, FAN X X, ZHU Z W, JIANG H B, WANG J, FAN Y C, XIONG C L, ZHENG Y Z, FU Z M, XU G J, CHEN D F, GUO R. Construction and annotation of the full-length transcriptome of Nosema ceranae based on the third-generation nanopore sequencing technology. Acta Entomologica Sinica, 2020,63(12):1461-1472. (in Chinese) | |
| [20] | 陈华枝, 范小雪, 范元婵, 王杰, 祝智威, 蒋海宾, 张文德, 隆琦, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 陈大福, 郭睿. 东方蜜蜂微孢子虫基因的可变剪接及可变腺苷酸化解析. 菌物学报, 2021,40(1):161-173. |
| CHEN H Z, FAN X X, FAN Y C, WANG J, ZHU Z W, JIANG H B, ZHANG W D, LONG Q, XIONG C L, ZHENG Y Z, FU Z M, XU G J, CHEN D F, GUO R. Analysis of alternative splicing and alternative polyadenylation of Nosema ceranae genes. Mycosystema, 2021,40(1):161-173. (in Chinese) | |
| [21] |
THIEL T, MICHALEK W, VARSHNEY R, GRANER A. Exploiting EST databases for the development and characterization of gene- derived SSR-markers in barley (Hordeum vulgare L.). Theoretical and Applied Genetics, 2003,106(3):411-422.
pmid: 12589540 |
| [22] | 郭睿, 陈华枝, 童新宇, 熊翠玲, 郑燕珍, 付中民, 解彦玲, 王海朋, 赵红霞, 陈大福. 蜜蜂球囊菌基因结构优化及新基因鉴定. 中国农业大学学报, 2019,24(1):61-68. |
| GUO R, CHEN H Z, TONG X Y, XIONG C L, ZHENG Y Z, FU Z M, XIE Y L, WANG H P, ZHAO H X, CHEN D F. Structural optimization of annotated genes and identification of novel genes in Ascosphaera apis. Journal of China Agricultural University, 2019,24(1):61-68. (in Chinese) | |
| [23] | 熊翠玲, 王海朋, 郑燕珍, 付中民, 徐国均, 童新宇, 赵红霞, 陈大福, 郭睿. 基于中华蜜蜂幼虫肠道转录组数据对东方蜜蜂基因组的基因结构优化及新基因鉴定. 中国农业大学学报, 2019,24(3):86-93. |
| XIONG C L, WANG H P, ZHENG Y Z, FU Z M, XU G J, TONG X Y, ZHAO H X, CHEN D F, GUO R. Gene structure optimization and identification of novel genes in Apis cerana genome: Based on the transcriptome data obtained from larval gut. Journal of China Agricultural University, 2019,24(3):86-93. (in Chinese) | |
| [24] |
CHENG B, FURTADO A, HENRY R J. Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Giga Science, 2017,6(11):1-13.
doi: 10.1093/gigascience/gix093 pmid: 29048480 |
| [25] |
GUO R, CHEN D F, XIONG C L, HOU C S, ZHENG Y Z, FU Z M, LIANG Q, DIAO Q Y, ZHANG L, WANG H Q, HOU Z X, KUMAR D. First identification of long non-coding RNAs in fungal parasite Nosema ceranae. Apidologie, 2018,49(5):660-670.
doi: 10.1007/s13592-018-0593-z |
| [26] |
BARRETT L W, FLETCHER S, WILTON S D. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cellular and Molecular Life Sciences, 2012,69(21):3613-3634.
pmid: 22538991 |
| [27] | 熊翠玲, 童新宇, 陈华枝, 耿四海, 庄天艺, 郑燕珍, 付中民, 陈大福, 赵红霞, 郭睿. 东方蜜蜂微孢子虫的基因结构优化及新基因鉴定. 环境昆虫学报, 2019,41(2):373-379. |
| XIONG C L, TONG X Y, CHEN H Z, GENG S H, ZHUANG T Y, ZHENG Y Z, FU Z M, CHEN D F, ZHAO H X, GUO R. Optimization of gene structure and identification of novel genes in Nosema ceranae. Journal of Environmental Entomology, 2019,41(2):373-379. (in Chinese) | |
| [28] |
JARNE P, LAGODA P J. Microsatellites, from molecules to populations and back. Trends in Ecology and Evolution, 1996,11(10):424-429.
pmid: 21237902 |
| [29] |
ZANE L, BARGELLONI L, PATARNELLO T. Strategies for microsatellite isolation: A review. Molecular Ecology, 2002,11(1):1-16.
doi: 10.1046/j.0962-1083.2001.01418.x pmid: 11903900 |
| [30] | 朱家颖, 吴国星, 杨斌. 基于转录组数据高通量发掘黄粉甲微卫星引物. 昆虫学报, 2013,56(7):724-728. |
| ZHU J Y, WU G X, YANG B. High-throughput discovery of SSR genetic markers in the yellow mealworm beetle, Tenebrio molitor (Coleoptera: Tenebrionidae), from its transcriptome database. Acta Entomologica Sinica, 2013,56(7):724-728. (in Chinese) | |
| [31] | 罗梅, 张鹤, 宾淑英, 林进添. 基于转录组数据高通量发掘扶桑绵粉蚧微卫星引物. 昆虫学报, 2014,57(4):395-400. |
| LUO M, ZHANG H, BIN S Y, LIN J T. High-throughput discovery of SSR genetic markers in the mealybug, Phenacoccus solenopsis (Hemiptera: Pseudococcidae), from its transcriptome database. Acta Entomologica Sinica, 2014,57(4):395-400. (in Chinese) | |
| [32] | 李汶东, 熊翠玲, 王鸿权, 侯志贤, 童新宇, 张璐, 付中民, 郑燕珍, 陈大福, 郭睿. 基于RNA-seq数据大规模挖掘蜜蜂球囊菌的SSR分子标记. 福建农林大学学报(自然科学版), 2017,46(4):434-438. |
| LI W D, XIONG C L, WANG H Q, HOU Z X, TONG X Y, ZHANG L, FU Z M, ZHENG Y Z, CHEN D F, GUO R. Large scale development of SSR molecular markers of Ascosphaera apis based on RNA-seq data. Journal of Fujian Agriculture and Forestry University (Natural Science Edition), 2017,46(4):434-438. (in Chinese) | |
| [33] | 熊翠玲, 张璐, 付中民, 王鸿权, 侯志贤, 童新宇, 李汶东, 郑燕珍, 陈大福, 郭睿. 基于RNA-seq数据大规模开发中华蜜蜂幼虫的SSR分子标记. 环境昆虫学报, 2017,39(1):68-74. |
| XIONG C L, ZHANG L, FU Z M, WANG H Q, HOU Z X, TONG X Y, LI W D, ZHENG Y Z, CHEN D F, GUO R. Large-scale development of SSR primers for Apis cerana cerana larvae based on its RNA-seq datasets. Journal of Environmental Entomology, 2017,39(1):68-74. (in Chinese) | |
| [34] | 郭睿, 陈华枝, 庄天艺, 熊翠玲, 郑燕珍, 付中民, 陈恒, 陈大福. 利用转录组数据开发意大利蜜蜂的SSR分子标记. 安徽农业大学学报, 2018,45(3):404-408. |
| GUO R, CHEN H Z, ZHUANG T Y, XIONG C L, ZHENG Y Z, FU Z M, CHEN H, CHEN D F. Exploitation of SSR markers for Apis mellifera ligustica based on transcriptome data. Journal of Anhui Agricultural University, 2018,45(3):404-408. (in Chinese) | |
| [35] | 张鹏飞, 周晓榕, 庞保平, 谭瑶, 常静, 高利军. 基于转录组数据高通量发掘沙葱萤叶甲微卫星引物. 应用昆虫学报, 2016,53(5):1058-1064. |
| ZHANG P F, ZHOU X R, PANG B P, TAN Y, CHANG J, GAO L J. High-throughput discovery of microsatellite markers in Galeruca daurica (Coleoptera: Chrysomelidae) from a transcriptome database. Chinese Journal of Applied Entomology, 2016,53(5):1058-1064. (in Chinese) |
| [1] | 叶美金, 吴雷, Lohani Md Nahibuzzaman, 尹丽, 胡欣荣, 刘亚西, 蒋云峰, 陈国跃, 蒲至恩, 李阳, 李婷, 邹亚亚, 吴佳怡, 马建. 基于GWAS的中国地方小麦成熟胚大小位点的鉴定及其遗传效应解析[J]. 中国农业科学, 2026, 59(6): 1157-1171. |
| [2] | 杨丽娟, 陈丝雨, 赵薇, 朱玲, 郭磊, 马丽娜, 马瑞敏, 张娟. 全基因组重测序揭示静原鸡羽色的遗传机制[J]. 中国农业科学, 2026, 59(6): 1348-1360. |
| [3] | 王勇胜, 牛丽, 王长杰, 马立花, 廉潇潇, 孟亚雄, 马小乐, 姚立蓉, 张宏, 杨轲, 李葆春, 王化俊, 司二静, 汪军成. 冬小麦千粒重的全基因组关联分析及候选基因预测[J]. 中国农业科学, 2026, 59(3): 499-514. |
| [4] | 贾子成, 秦冰雨, 马彩英, 杜勇, 刘统高, 薛瑞林, 王小龙, 周世卫. 饲粮营养水平对双羔陕北白绒山羊母子一体化生产性能和瘤胃微生物的影响[J]. 中国农业科学, 2026, 59(3): 668-686. |
| [5] | 王忠妮, 雷月, 李佳丽, 宫彦龙, 朱速松. ABC转运蛋白OsARG1调控水稻抽穗期的功能[J]. 中国农业科学, 2026, 59(1): 1-16. |
| [6] | 王思琪, 邹利人, 白瑞雯, 闫可, 王思洋, 齐晓光, 申海林, 温景辉. 赤霉素调控‘蜜汁’葡萄穗轴硬化关键基因的挖掘[J]. 中国农业科学, 2026, 59(1): 179-189. |
| [7] | 李云丽, 刁邓超, 刘雅睿, 孙玉晨, 孟祥宇, 邬陈芳, 汪妤, 吴建辉, 李春莲, 曾庆东, 韩德俊, 郑炜君. 小麦苗期耐热性全基因组关联分析[J]. 中国农业科学, 2025, 58(9): 1663-1683. |
| [8] | 张义茹, 韩雪, 姚鑫杰, 冯军, 魏爱丽, 李文超, 张彬, 韩渊怀, 李红英. 基于多组学解析谷子后熟米色变化的分子机制[J]. 中国农业科学, 2025, 58(9): 1702-1718. |
| [9] | 邹晓威, 夏蕾, 朱晓敏, 孙辉, 周琦, 齐霁, 张亚封, 郑岩, 姜兆远. 基于转录组测序的玉米瘤黑粉菌UM01240过表达菌株诱导玉米抗病性分析[J]. 中国农业科学, 2025, 58(6): 1116-1130. |
| [10] | 孙萍, 朱文灿, 林贤锐, 吴嘉颀, 曹译文, 陈辰斐, 王轶, 朱建锡, 贾惠娟, 钱敏杰, 沈建生. 基于代谢组和转录组解析多雨寡照对桃果皮着色和类黄酮积累的影响[J]. 中国农业科学, 2025, 58(6): 1173-1194. |
| [11] | 谢露露, 李福, 张思远, 高建昌. 基于跨物种转录组解析影响不定根发生的保守基因[J]. 中国农业科学, 2025, 58(6): 1195-1209. |
| [12] | 高岩浩, 王婷婷, 白卫卫, 杜兴杰, 刘贤, 秦本源, 付彤, 孙宇, 高腾云, 张天留. 脂质组与转录组联合揭示南阳牛不同肌肉组织脂质特征的差异表达模式[J]. 中国农业科学, 2025, 58(6): 1239-1258. |
| [13] | 罗正英, 胡鑫, 吴转娣, 钱禛锋, 田春艳, 刘新龙, 李富生. 蔗茅全基因组SSR标记特征分析与开发[J]. 中国农业科学, 2025, 58(5): 851-863. |
| [14] | 张天雨, 李白, 藏金萍, 曹宏哲, 董金皋, 邢继红, 张康. 灰葡萄孢HMG家族基因的全基因组鉴定与表达规律分析[J]. 中国农业科学, 2025, 58(4): 704-718. |
| [15] | 周广飞, 马亮, 马璐, 张舒钰, 章慧敏, 宋旭东, 张振良, 陆虎华, 郝德荣, 冒宇翔, 薛林, 陈国清. 玉米苞叶性状全基因组关联分析[J]. 中国农业科学, 2025, 58(3): 431-442. |
|
||