






中国农业科学 ›› 2019, Vol. 52 ›› Issue (1): 166-180.doi: 10.3864/j.issn.0578-1752.2019.01.015
郭睿( ),杜宇(),童新宇,熊翠玲,郑燕珍,徐国钧,王海朋,耿四海,周丁丁,郭意龙,吴素珍,陈大福()
),杜宇(),童新宇,熊翠玲,郑燕珍,徐国钧,王海朋,耿四海,周丁丁,郭意龙,吴素珍,陈大福()
收稿日期:2018-08-02
接受日期:2018-10-01
出版日期:2019-01-01
发布日期:2019-01-12
基金资助:
GUO Rui(),DU Yu(),TONG XinYu,XIONG CuiLing,ZHENG YanZhen,XU GuoJun,WANG HaiPeng,GENG SiHai,ZHOU DingDing,GUO YiLong,WU SuZhen,CHEN DaFu()
Received:2018-08-02
Accepted:2018-10-01
Published:2019-01-01
Online:2019-01-12
摘要:
【目的】 微小RNA(microRNA,miRNA)是一类重要的基因表达调控因子,可影响宿主与病原间的互作过程。蜜蜂球囊菌(Ascosphaera apis)是一种特异性侵染蜜蜂幼虫的致死性真菌病原。本研究旨在对意大利蜜蜂(Apis mellifera ligustica,简称意蜂)幼虫肠道在球囊菌胁迫前期的差异表达miRNA(differentially expressed miRNA,DEmiRNA)及其靶基因进行深入分析,在miRNA组学水平探究意蜂幼虫在球囊菌侵染前期的胁迫应答,并通过构建显著DEmiRNA的调控网络筛选出与宿主应答相关的关键miRNA。【方法】 利用small RNA-seq(sRNA-seq)技术对正常及球囊菌侵染的意蜂4日龄幼虫肠道(AmCK和AmT)进行高通量测序,首先对原始数据进行质控和评估,随后将过滤后的数据与西方蜜蜂(Apis mellifera)参考基因组进行比对;将比对上的序列标签(tags)注释到miRBase数据库,得出已知miRNA的表达量;通过TPM(tags per million)算法对各样本中miRNA的表达量进行归一化处理,以|log2 fold change|≥1且P≤0.05作为标准筛选得到显著DEmiRNA;利用TargetFinder 软件预测显著DEmiRNA的靶基因,并对其进行GO和KEGG代谢通路(pathway)富集分析。利用Cytoscape软件对miRNA-mRNA调控网络进行可视化。最后,利用茎环反转录PCR(Stem-loop RT-PCR)和荧光定量PCR(qPCR)验证测序数据的可靠性。【结果】 AmCK和AmT样品的测序分别得到13 553 302和10 777 534条原始读段(raw reads),经严格过滤后得到的有效读段(clean reads)数分别为13 186 921和10 480 913条。各样品的生物学重复间的Pearson相关性系数分别在0.9822和0.9508以上。共有10个显著DEmiRNA,包括4个上调miRNA和6个下调miRNA。显著DEmiRNA在AmT的整体表达水平低于AmCK。10个显著DEmiRNA可靶向结合3 788个靶基因,其中上调miRNA的1 240个靶基因可注释到GO数据库中的39个GO条目,主要富集在结合、细胞进程、代谢进程和应激反应等;下调miRNA的749个靶基因可注释到34个GO条目,主要富集在细胞进程、结合、代谢进程和应激反应等。KEGG数据库注释结果显示,上调miRNA和下调miRNA的靶基因分别注释到95和66条代谢通路,富集基因数最多的分别是Wnt信号通路、Hippo信号通路、光传导以及内吞作用、磷脂酰肌醇信号系统、嘌呤代谢。对于上调和下调miRNA,分别有31和52个靶基因注释到内吞作用,15和7个靶基因注释到泛素介导的蛋白水解,11和5个靶基因注释到Jak-STAT信号通路,1和3个靶基因注释到MAPK信号通路。显著DEmiRNA与靶mRNA之间形成复杂的调控网络,7个显著DEmiRNA靶向结合96个与Wnt信号通路相关的mRNA,8个显著DEmiRNA靶向结合55个与内吞作用相关的mRNA。Stem-loop RT-PCR和qPCR结果验证了测序数据的可靠性。【结论】 对意蜂幼虫肠道在球囊菌侵染前期的DEmiRNA及其靶基因进行预测和分析,并构建和分析了DEmiRNA-mRNA调控网络,研究结果提供了宿主miRNA的表达谱和差异表达信息,揭示了DEmiRNA通过调控细胞生命活动、新陈代谢以及部分细胞和体液免疫等生物学过程参与宿主的胁迫应答。miR-4331-y、miR-4968-y、miR-8440-y、novel-m0023-5p和novel-m0025-3p共同参与了宿主的Wnt信号通路和内吞作用的调控,可作为白垩病治疗的潜在分子靶标。
郭睿,杜宇,童新宇,熊翠玲,郑燕珍,徐国钧,王海朋,耿四海,周丁丁,郭意龙,吴素珍,陈大福. 意大利蜜蜂幼虫肠道在球囊菌侵染前期的 差异表达microRNA及其调控网络[J]. 中国农业科学, 2019, 52(1): 166-180.
GUO Rui,DU Yu,TONG XinYu,XIONG CuiLing,ZHENG YanZhen,XU GuoJun,WANG HaiPeng,GENG SiHai,ZHOU DingDing,GUO YiLong,WU SuZhen,CHEN DaFu. Differentially Expressed MicroRNAs and Their Regulation Networks in Apis mellifera ligustica Larval Gut During the Early Stage of Ascosphaera apis Infection[J]. Scientia Agricultura Sinica, 2019, 52(1): 166-180.
表1
本研究使用的引物"
| 引物名称 Primer name | 引物序列 Primer sequence |
|---|---|
| LOOP-miR-3793-x | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGGCCAGG |
| LOOP-ame-miR-6000a-5p | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATAGAGAC |
| LOOP-novel-m0031-3p | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCCTGCTT |
| LOOP-novel-m0034-5p | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATATCACA |
| F-miR-3793-x | ACACTCCAGCTGGGAGCGTGTTTTC |
| F-ame-miR-6000a-5p | ACACTCCAGCTGGGCAGCAGCAGCAG |
| F-novel-m0031-3p | GCATCCTCTTGAAT |
| F-novel-m0034-5p | CAGGTAACTACTGC |
| R | CTCAACTGGTGTCGTGGA |
| U6-F | GTTAGGCTTTGACGATTTCG |
| U6-R | GGCATTTCTCCACCAGGTA |
表2
sRNA-seq数据总览"
| 样品Sample | 原始读段Raw reads | 有效读段Clean reads |
|---|---|---|
| AmCK-1 | 15146178 | 14736731 (97.30%) |
| AmCK-2 | 13310167 | 12954535 (97.33%) |
| AmCK-3 | 12203560 | 11869496 (97.26%) |
| AmT-1 | 10741447 | 10453795 (97.32%) |
| AmT-2 | 12035887 | 11694678 (97.17%) |
| AmT-3 | 9555268 | 9294267 (97.27%) |
表3
AmCK vs AmT 中DEmiRNA的信息统计"
| 差异表达miRNA ID DEmiRNA ID | AmCK中的表达量 Expression in AmCK | AmT中的表达量 Expression in AmT | 以2为底miRNA的相对变化倍数的对数值 log2 fold change | 靶基因数 Number of target genes |
|---|---|---|---|---|
| miR-7085-x | 0.01 | 8.40 | 9.71 | 129 |
| miR-8440-y | 0.01 | 5.29 | 9.05 | 2265 |
| novel-m0023-5p | 0.01 | 1.79 | 7.48 | 334 |
| miR-3793-x | 33.72 | 89.76 | 1.41 | 398 |
| novel-m0034-3p | 4.06 | 1.95 | -1.05 | 768 |
| miR-4331-y | 74.07 | 27.70 | -1.42 | 77 |
| ame-miR-6000a-5p | 19.06 | 4.14 | -2.20 | 77 |
| miR-971-y | 3.25 | 0.31 | -3.37 | 100 |
| novel-m0025-3p | 0.88 | 0.01 | -6.46 | 294 |
| miR-4968-y | 11.61 | 0.01 | -10.18 | 279 |
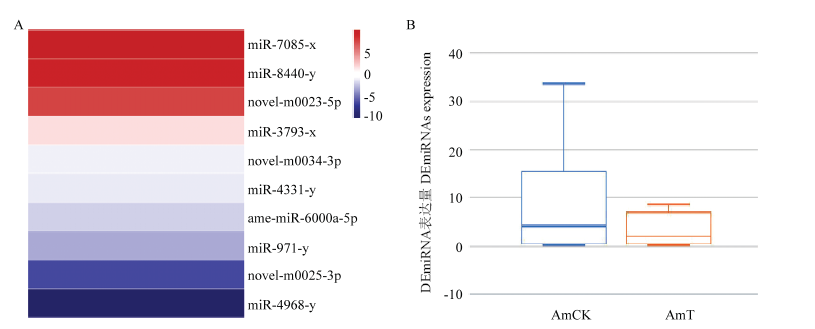
图1
AmCK vs AmT中DEmiRNA的表达谱 A:DEmiRNA的表达量聚类Expression clustering of DEmiRNAs;B:不同样品中DEmiRNA的表达量比较Comparison of DEmiRNAs expression in different samples"
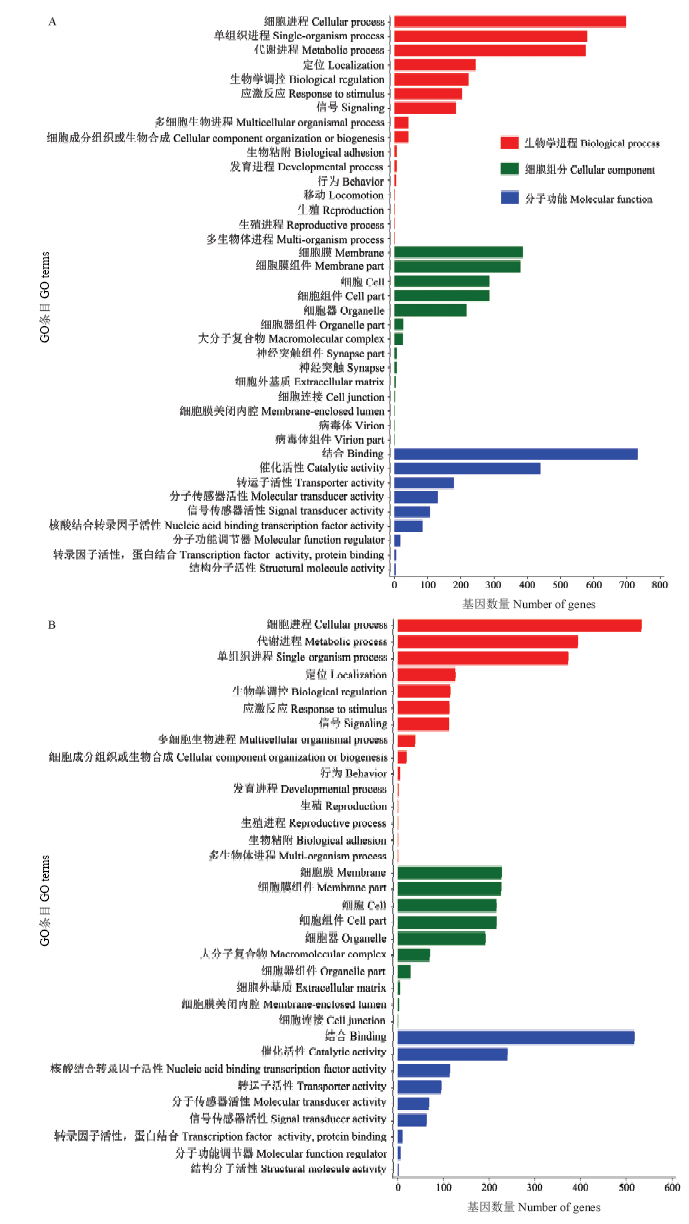
图2
AmCK vs AmT中显著DEmiRNA的靶基因的GO数据库注释 A:上调miRNA的靶基因Target genes of up-regulated miRNAs;B:下调miRNA的靶基因Target genes of down-regulated miRNAs"

图3
AmCK vs AmT中DEmiRNA的靶基因的KEGG数据库注释 A:上调miRNA的靶基因Target genes of up-regulated miRNAs;B:下调miRNA的靶基因Target genes of down-regulated miRNAs"
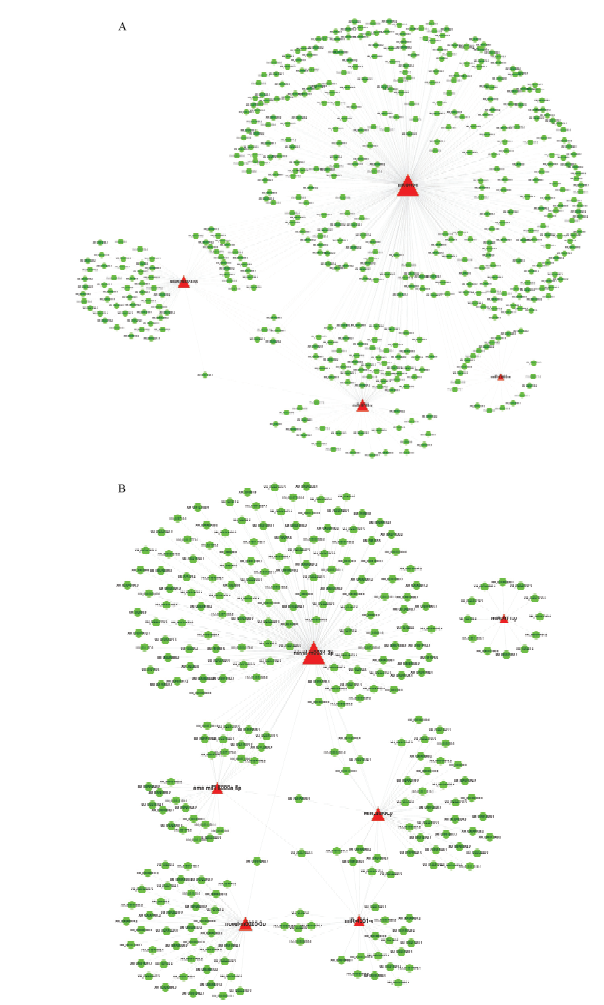
图4
意蜂幼虫肠道的DEmiRNA的调控网络 A: 上调miRNA的调控网络Regulation networks of up-regulated miRNAs; B:下调miRNA的调控网络Regulation networks of down-regulated miRNAs。绿色圆形代表靶基因,红色三角代表miRNA Green circles indicate target genes, red triangles indicate miRNAs"
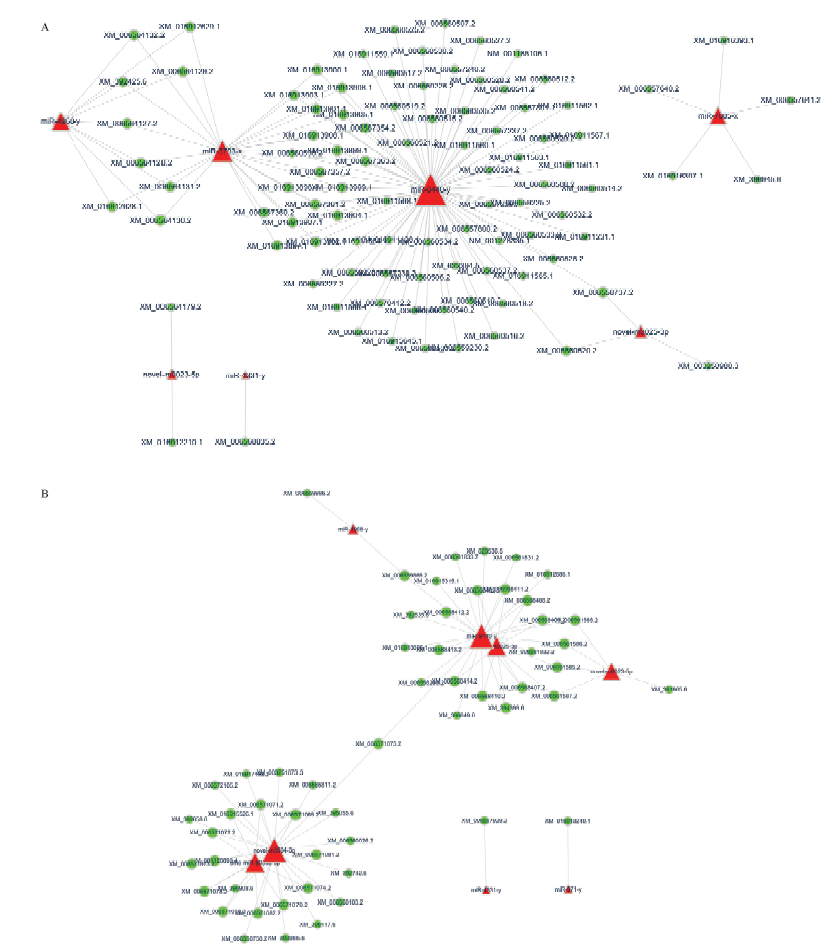
图5
意蜂幼虫肠道的Wnt信号通路和内吞作用相关显著DEmiRNA的调控网络 A: Wnt信号通路相关DEmiRNA的调控网络Regulation networks of DEmiRNAs related to Wnt signaling pathway; B:内吞作用相关DEmiRNA的调控网络Regulation networks of DEmiRNAs related to endocytosis。绿色圆形代表靶基因,红色三角代表miRNA Green circles indicate target genes, red triangles indicate miRNAs"
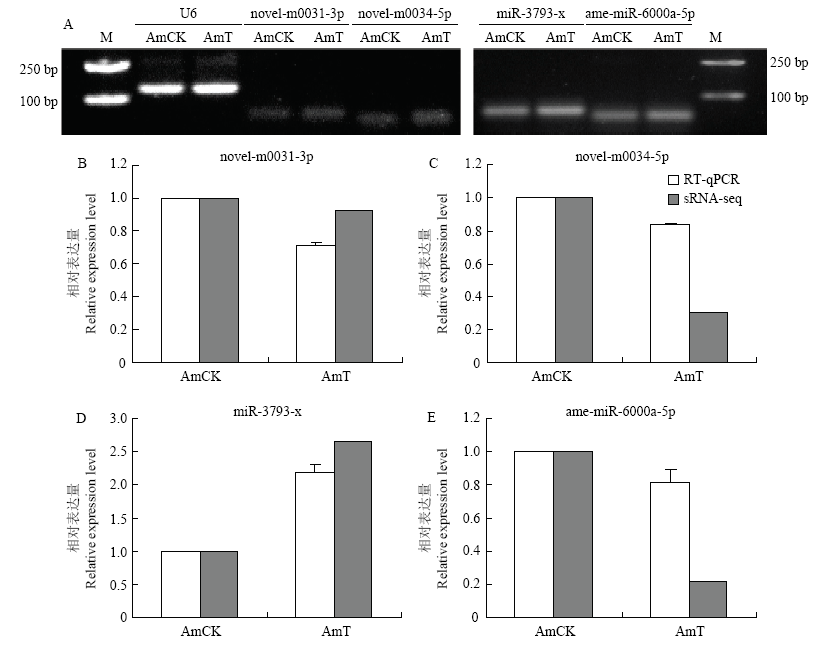
图6
DEmiRNA的Stem-loop RT-PCR(A)和qPCR验证(B—E)"
| [1] |
罗术东, 王彪, 褚忠桥, 柳萌, 吴杰 . 不同蜂为设施辣椒授粉的授粉效果比较. 环境昆虫学报, 2015,37(2):381-386.
doi: 10.3969/j.issn.1674-0858.2015.02.23 |
|
LUO S D, WANG B, CHU Z Q, LIU M, WU J . Comparison of the pollination effects for pepper between different bees in greenhouse. Journal of Environmental Entomology, 2015,37(2):381-386. (in Chinese)
doi: 10.3969/j.issn.1674-0858.2015.02.23 |
|
| [2] | 李江红, 郑志阳, 陈大福, 梁勤 . 影响蜜蜂球囊菌侵染蜜蜂幼虫的因素及侵染过程观察. 昆虫学报, 2012,55(7):790-797. |
| LI J H, ZHENG Z Y, CHEN D F, LIANG Q . Factors influencing Ascosphaera apis infection on honeybee larvae and observation on the infection process. Acta Entomologica Sinica, 2012,55(7):790-797. (in Chinese) | |
| [3] |
ASGARI S . MicroRNA functions in insects. Insect Biochemistry and Molecular Biology, 2013,43(4):388-397.
doi: 10.1016/j.ibmb.2012.10.005 pmid: 23103375 |
| [4] |
CHEN J F, MANDEL E M, THOMSON J M, WU Q, CALLIS T E, HAMMOND S M, CONLON F L, WANG D Z . The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nature Genetics, 2006,38(2):228-233.
doi: 10.1038/ng1725 pmid: 2538576 |
| [5] |
ALVAREZ-GARCIA I, MISKA E A . MicroRNA functions in animal development and human disease. Development, 2005,132(21):4653-4662.
doi: 10.1242/dev.02073 pmid: 16224045 |
| [6] |
GUO C J, PAN Q, LI D G, SUN H, LIU B W . MiR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: An essential role for apoptosis. Journal of Hepatology, 2009,50(4):766-778.
doi: 10.1016/j.jhep.2008.11.025 pmid: 19232449 |
| [7] |
SCARIA V, HARIHARAN M, MAITI S, PILLAI B, BRAHMACHARI S K . Host-virus interaction: A new role for microRNAs. Retrovirology, 2006,3:68.
doi: 10.1186/1742-4690-3-68 pmid: 17032463 |
| [8] | LI S, SHEN L, SUN L, XU J, JIN P, CHEN L, MA F . Small RNA-Seq analysis reveals microRNA-regulation of the Imd pathway during Escherichia coli infection in Drosophila. Developmental and Comparative Immunology, 2017,70:80-87. |
| [9] | QIAN P, JIANG T, WANG X, SONG F, CHEN C, SHEN X . Bmo-miR-275 down-regulates expression of Bombyx mori sericin gene 2 in vitro. PLoS ONE, 2018,13(1):e0190464. |
| [10] |
ZHANG G, HUSSAIN M , O’NEILL S L, ASGARI S. Wolbachia uses a host microRNA to regulate transcripts of a methyltransferase, contributing to dengue virus inhibition in Aedes aegypti. Proceedings of the National Academy of Sciences of the United States of America, 2013,110(25):10276-10281.
doi: 10.1073/pnas.1303603110 pmid: 23733960 |
| [11] | 李盛杰 . microRNA在果蝇Toll信号免疫响应中的调控作用研究[D]. 南京: 南京师范大学, 2017. |
| LI S J . Regulation of microRNA on Toll signal immune response in Drosophila melanogaster[D]. Nanjing: Nanjing Normal University, 2017. ( in Chinese) | |
| [12] |
LOURENÇO A P, GUIDUGLILAZZARINI K R, FREITAS F C, BITONDI M M, SIMÕES Z L . Bacterial infection activates the immune system response and dysregulates microRNA expression in honey bees. Insect Biochemistry and Molecular Biology, 2013,43(5):474-482.
doi: 10.1016/j.ibmb.2013.03.001 |
| [13] |
HUANG Q, CHEN Y P, RUI W W, SCHWARZ R S , EVANS J D. Honeybee microRNAs respond to infection by the microsporidian parasite Nosema ceranae. Scientific Reports, 2015, 5: Article number 17494.
doi: 10.1038/srep17494 pmid: 26620304 |
| [14] |
陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 张曌楠, 黄枳腱, 张璐, 王鸿权, 解彦玲, 童新宇 . 中华蜜蜂幼虫肠道响应球囊菌早期胁迫的转录组学. 中国农业科学, 2017,50(13):2614-2623.
doi: 10.3864/j.issn.0578-1752.2017.13.019 |
|
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, ZHANG Z N, HUANG Z J, ZHANG L, WANG H Q, XIE Y L, TONG X Y . Transcriptome of Apis cerana cerana larval gut under the stress of Ascosphaera apis. Scientia Agricultura Sinica, 2017,50(13):2614-2623. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2017.13.019 |
|
| [15] |
郭睿, 熊翠玲, 郑燕珍, 张璐, 童新宇, 梁勤, 陈大福 . 意大利蜜蜂幼虫肠道响应球囊菌早期胁迫的转录组学分析. 应用昆虫学报, 2017,54(4):553-560.
doi: 10.7679/j.issn.2095-1353.2017.067 |
|
GUO R, XIONG C L, ZHENG Y Z, ZHANG L, TONG X Y, LIANG Q, CHEN D F . Transcriptome analysis of Apis mellifera ligustica larval gut during the early stage of stress induced by Ascosphaera apis. Chinese Journal of Applied Entomology, 2017,54(4):553-560. (in Chinese)
doi: 10.7679/j.issn.2095-1353.2017.067 |
|
| [16] |
HUSSAIN M, ASGARI S . MicroRNAs as mediators of insect host-pathogen interactions and immunity. Journal of Insect Physiology, 2014,70:151-158.
doi: 10.1016/j.jinsphys.2014.08.003 pmid: 25152509 |
| [17] | 郭睿, 王海朋, 陈华枝, 熊翠玲, 郑燕珍, 付中民, 赵红霞, 陈大福 . 蜜蜂球囊菌的microRNA鉴定及其调控网络分析. 微生物学报, 2018,58(6):1077-1089. |
| GUO R, WANG H P, CHEN H Z, XIONG C L, ZHENG Y Z, FU Z M, ZHAO H X, CHEN D F . Identification of Ascosphaera apis microRNAs and investigation of their regulation networks. Acta Microbiologica Sinica, 2018,58(6):1077-1089. (in Chinese) | |
| [18] |
FRIEDLÄNDER M R, MACKOWIAK S D, LI N, CHEN W, RAJEWSKY N . MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Research, 2012,40(1):37-52.
doi: 10.1093/nar/gkr688 pmid: 21911355 |
| [19] |
ALLEN E, XIE Z, GUSTAFSON A M, CARRINGTON J C . MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell, 2005,121(2):207-221.
doi: 10.1016/j.cell.2005.04.004 pmid: 15851028 |
| [20] |
CHEN C, RIDZON D A, BROOMER A J, LEE D H, NGUYEN J T, BARBISIN M, XU N L, MAHUVAKAR V R, ANDERSEN M R, LAO K Q, LIVAK K J, GUEGLER K J . Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Research, 2005,33(20):e179.
doi: 10.1093/nar/gni178 |
| [21] |
赵红霞, 梁勤, 罗岳雄, 李江红, 张学锋, 曾鑫年 . 蜜蜂白垩病的研究进展. 环境昆虫学报, 2014,36(2):233-239.
doi: 10.3969/j.issn.1674-0858.2014.02.18 |
|
ZHAO H X, LIANG Q, LUO Y X, LI J H, ZHANG X F, ZENG X N . Chalkbrood disease in honeybee. Journal of Environmental Entomology, 2014,36(2):233-239. (in Chinese)
doi: 10.3969/j.issn.1674-0858.2014.02.18 |
|
| [22] |
ZHAO L, ZHU J, ZHOU H, ZHAO Z, ZOU Z, LIU X, LIN X, ZHANG X, DENG X, WANG R, CHEN H , JIN M. Identification of cellular microRNA-136 as a dual regulator of RIG-I-mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Scientific Reports, 2015, 5: Article number 14991.
doi: 10.1038/srep14991 pmid: 4598873 |
| [23] |
HU Y, JIANG L, LAI W, QIN Y, ZHANG T, WANG S, YE X . MicroRNA-33a disturbs influenza A virus replication by targeting ARCN1 and inhibiting viral ribonucleoprotein activity. The Journal of General Virology, 2016,97(1):27-38.
doi: 10.1099/jgv.0.000311 pmid: 26498766 |
| [24] | CHEN D, GUO R, XU X, XIONG C, LIANG Q, ZHENG Y, LUO Q, ZHANG Z, HUANG Z, KUMAR D, XI W, ZOU X, LIU M . Uncovering the immune responses of Apis mellifera ligustica larval gut to Ascosphaera apis infection utilizing transcriptome sequencing. Gene, 2017,621:40-50. |
| [25] |
陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 黄枳腱, 张曌楠, 张璐, 李汶东, 童新宇, 席伟军 . 胁迫意大利蜜蜂幼虫肠道的球囊菌的转录组分析. 昆虫学报, 2017,60(4):401-411.
doi: 10.16380/j.kcxb.2017.04.005 |
|
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, HUANG Z J, ZHANG Z N, ZHANG L, LI W D, TONG X Y, XI W J . Transcriptomic analysis of Ascosphaera apis stressing larval gut of Apis mellifera ligustica( Hyemenoptera: Apidae) . Acta Entomologica Sinica, 2017,60(4):401-411. (in Chinese)
doi: 10.16380/j.kcxb.2017.04.005 |
|
| [26] |
郭睿, 陈大福, 黄枳腱, 梁勤, 熊翠玲, 徐细建, 郑燕珍, 张曌楠, 解彦玲, 童新宇, 候志贤, 江亮亮, 刀晨 . 球囊菌胁迫中华蜜蜂幼虫肠道过程中病原的转录组学研究. 微生物学报, 2017,57(12):1865-1878.
doi: 10.13343/j.cnki.wsxb.20160551 |
|
GUO R, CHEN D F, HUANG Z J, LIANG Q, XIONG C L, XU X J, ZHENG Y Z, ZHANG Z N, XIE Y L, TONG X Y, HOU Z X, JIANG L L, DAO C . Transcriptome analysis of Ascosphaera apis stressing larval gut of Apis cerana cerana. Acta Microbiologica Sinica, 2017,57(12):1865-1878. (in Chinese)
doi: 10.13343/j.cnki.wsxb.20160551 |
|
| [27] | AYYAZ A, LI H, JASPER H . Hemocytes control stem cell activity in the Drosophila intestine. Nature Cell Biology, 2015,17(6):736-748. |
| [28] |
BARRY E R, CAMARGO F D . The Hippo superhighway: signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Current Opinion in Cell Biology, 2013,25(2):247-253.
doi: 10.1016/j.ceb.2012.12.006 pmid: 23312716 |
| [29] | 郭睿, 耿四海, 熊翠玲, 郑燕珍, 付中民, 王海朋, 杜宇, 童新宇, 赵红霞, 陈大福 . 意大利蜜蜂工蜂中肠发育过程中长链非编码RNA的差异表达分析. 中国农业科学, 2018,51(18):3600-3613. |
| GUO R, GENG S H, XIONG C L, ZHENG Y Z, FU Z M, WANG H P, DU Y, TONG X Y, ZHAO H X, CHEN D F . Differential expression analysis of long non-coding RNAs during the developmental process of Apis mellifera ligustica worker’s midgut. Scientia Agricultura Sinica, 2018,51(18):3600-3613. (in Chinese) | |
| [30] | 郭睿, 陈华枝, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 杜宇, 王海朋, 耿四海, 周丁丁, 刘思亚, 陈大福 . 意大利蜜蜂工蜂中肠发育过程中的差异表达环状RNA及其调控网络分析. 中国农业科学, 2018,51(23):4575-4590. |
| GUO R, CHEN H Z, XIONG C L, ZHENG Y Z, FU Z M, XU G J, DU Y, WANG H P, GENG S H, ZHOU D D, LIU S Y, CHEN D F . Analysis of differentially expressed circular RNAs and their regulation networks during the developmental process of Apis mellifera ligustica worker’s midgut. Scientia Agricultura Sinica, 2018,51(23):4575-4590. (in Chinese) | |
| [31] |
ASHBY R, FORÊT S, SEARLE I , MALESZKA R. MicroRNAs in honeybee caste determination. Scientific Reports, 2016, 6: Article number 18794.
doi: 10.1038/srep18794 pmid: 4704047 |
| [32] | 郝向伟 . 家蚕Hedgehog信号通路相关基因的克隆、鉴定及其功能分析[D]. 重庆: 西南大学, 2013. |
| HAO X W . Cloning, characterization and functional analysis of genes in the hedgehog signaling pathway in the intestine of silkworm, Bombyx mori[D]. Chongqing: Southwest University, 2013. ( in Chinese) | |
| [33] |
CHA Y H, KIM N H, PARK C, LEE I, KIM H S, YOOK J I . MiRNA-34 intrinsically links p53 tumor suppressor and Wnt signaling. Cell Cycle, 2012,11(7):1273-1281.
doi: 10.4161/cc.19618 pmid: 22421157 |
| [34] |
KIM N H, KIM H S, KIM N G, LEE I, CHOI H S, LI X Y, KANG S E, CHA S Y, RYU J K, NA J M, PARK C, KIM K, LEE S, GUMBINER B M, YOOK J I , WEISS S J. P53 and microRNA-34 are suppressors of canonical Wnt signaling. Science Signaling, 2011, 4(197): ra71.
doi: 10.1126/scisignal.2001744 pmid: 3447368 |
| [35] | 陈晓 . 蜜蜂卵巢激活和产卵过程差异表达的编码RNA与非编码RNA的筛选和鉴定[D]. 北京: 中国农业科学院, 2017. |
| CHEN X . Identification of differentially expressed coding and noncoding RNAs during ovary activation and oviposition in honeybees[D]. Beijing: Chinese Academy of Agricultural Sciences, 2017. ( in Chinese) | |
| [36] |
BARIL M, ES-SAAD S, CHATEL-CHAIX L, FINK K, PHAM T, RAYMOOD V A, AUDETTE K, GUENIER A S, DUCHAINE J, SERVANT M, BILODEAU M, COHEN E, GRANDVAUX N, LAMARRE D . Genome-wide RNAi screen reveals a new role of a Wnt/CTNNB1 signaling pathway as negative regulator of virus-induced innate immune responses. PLoS Pathogens, 2013,9(6):e1003416.
doi: 10.1371/journal.ppat.1003416 pmid: 3681753 |
| [37] |
HACK K, REILLY L, PROBY C, FLEMING C, LEIGH I, FOERSTER J . Wnt5a inhibits the CpG oligodeoxynucleotide- triggered activation of human plasmacytoid dendritic cells. Clinical and Experimental Dermatology, 2012,37(5):557-561.
doi: 10.1111/j.1365-2230.2012.04362.x pmid: 22607321 |
| [38] |
SMITH J L, JENG S, MCWEENEY S K, HIRSCH A J . A microRNA screen identifies the Wnt signaling pathway as a regulator of the interferon response during flavivirus infection. Journal of Virology, 2017,91(8):e02388-16.
doi: 10.1128/JVI.02388-16 pmid: 28148804 |
| [39] | 乔莹 . 刺激隐核虫感染大黄鱼的miRNA和mRNA组学测序及关联分析[D]. 厦门: 厦门大学, 2016. |
| QIAO Y . Sequencing and correlation analysis of miRNAome and transcriptome of Larimichthys crocea infected by Cryptocaryon irritans[D]. Xiamen: Xiamen University, 2016. ( in Chinese) | |
| [40] |
HE L, HANNON G J . MicroRNAs: small RNAs with a big role in gene regulation. Nature Reviews Genetics, 2004,5(7):522-531.
doi: 10.1038/nrg1379 pmid: 15211354 |
| [41] | 郭睿, 杜宇, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 王海朋, 陈华枝, 耿四海, 周丁丁, 石彩云, 赵红霞, 陈大福 . 意大利蜜蜂幼虫肠道发育过程中的差异表达microRNA及其调控网络. 中国农业科学, 2018,51(21):4197-4209. |
| GUO R, DU Y, XIONG C L, ZHENG Y Z, FU Z M, XU G J, WANG H P, CHEN H Z, GENG S H, ZHOU D D, SHI C Y, ZHAO H X, CHEN D F . Differentially expressed microRNA and their regulation networks during the developmental process of Apis mellifera ligustica larval gut. Scientia Agricultura Sinica, 2018,51(21):4197-4209. (in Chinese) | |
| [42] |
ZHAO X, BAI X, GUAN L, LI J, SONG X, MA X, GUO J, ZHANG Z, DU Q, HUANG Y, TONG D . MicroRNA-4331 promotes Transmissible Gastroenteritis Virus (TGEV)-induced mitochondrial damage via targeting RB1, upregulating interleukin-1 receptor accessory protein (IL1RAP), and activating p38 MAPK Pathway in vitro. Molecular and Cellular Proteomics, 2018,17(2):190-204.
doi: 10.1074/mcp.RA117.000432 |
| [43] |
WANG Y, BRAHMAKSHATRIYA V, LUPIANI B, REDDY S M, SOIBAM B, BENHAM A L, GUNARATNE P, LIU H C, TRAKOOLJUL N, ING N, OKIMOTO R, ZHOU H . Integrated analysis of microRNA expression and mRNA transcriptome in lungs of avian influenza virus infected broilers. BMC Genomics, 2012,13:278.
doi: 10.1186/1471-2164-13-278 pmid: 3496578 |
| [44] |
TAMBYAH P A, SEPRAMANIAM S, MOHAMED ALI J, CHAI S C, SWAINATHAN P, ARMUGAM A, JEYASEELAN K . MicroRNAs in circulation are altered in response to influenza A virus infection in humans. PLoS ONE, 2013,8(10):e76811.
doi: 10.1371/journal.pone.0076811 pmid: 24116168 |
| [45] |
ZHANG S, WANG R, SU H, WANG B, SIZHU S, LEI Z, JIN M, CHEN H, CAO J, ZHOU H . Sus scrofa miR-204 and miR-4331 negatively regulate swine H1N1/2009 influenza A virus replication by targeting viral HA and NS, respectively. International Journal of Molecular Sciences, 2017,18(4):E749.
doi: 10.3390/ijms18040749 pmid: 28368362 |
| [46] |
WANG P, GRANADOS R R . Molecular structure of the peritrophic membrane (PM): identification of potential PM target sites for insect control. Archives of Insect Biochemistry and Physiology, 2001,47(2):110-118.
doi: 10.1002/arch.1041 pmid: 11376457 |
| [1] | 潘丽媛, 王永军, 李海军, 侯富, 李菁, 李丽丽, 孙苏阳. 基于转录组和WGCNA筛选小麦籽粒蛋白质累积相关调控基因[J]. 中国农业科学, 2025, 58(6): 1065-1082. |
| [2] | 许朵朵, 杜倩倩, 赵理想, 栗燕, 黄淦, 李永华, 逯久幸. 牡丹AP2/ERF转录因子的全基因组分析[J]. 中国农业科学, 2025, 58(23): 5031-5045. |
| [3] | 吴东明, 徐靖, 袁佳美, 王珂璇, 李玉义, 何萍, 张建峰, 宋大利, 高淼, 周卫. 一株根际促生菌Myroides odoratimimus PJ-3的分离、鉴定及其耐盐碱和玉米促生性能评价[J]. 中国农业科学, 2025, 58(20): 4131-4143. |
| [4] | 张鑫瑶, 王萍, 刘亚龙, 汪景宽. 不同植稻年限土壤酶活性及其化学计量特征[J]. 中国农业科学, 2025, 58(13): 2604-2613. |
| [5] | 王慧玲, 张莹莹, 闫爱玲, 王晓玥, 刘振华, 任建成, 徐海英, 孙磊. 多组学联合解析红色玫瑰香型葡萄品种转色过程中单萜和花色苷积累规律[J]. 中国农业科学, 2025, 58(13): 2645-2662. |
| [6] | 周旗, 张亮, 潘雨, 涂志, 王峥, 刘航航, 鲜凌瑾, 夏运红, 潘红梅, 龙熙. 环黄芪醇对猪供体成纤维细胞衰老、细胞骨架与核移植胚胎早期发育的影响[J]. 中国农业科学, 2025, 58(12): 2453-2474. |
| [7] | 张余周, 王一钊, 高茹茜, 刘逸凡. 小麦根系构型及抗旱性研究进展[J]. 中国农业科学, 2024, 57(9): 1633-1645. |
| [8] | 张必东, 林泓, 朱思颖, 李忠成, 庄慧, 李云峰. 水稻颖壳异常突变体ah1的鉴定与候选基因分析[J]. 中国农业科学, 2024, 57(3): 429-441. |
| [9] | 池润清, 韩海银, 王鹏, 李凯扬, 储明星, 刘玉芳. 雌激素介导circZNF423作为ceRNA调控oar-miR-541-3p/CALM3通路促进绵羊成肌细胞增殖[J]. 中国农业科学, 2024, 57(3): 597-612. |
| [10] | 曾祥翠, 杨永念, 李如月, 蒋学乾, 蒋旭, 徐嫣然, 刘忠宽, 龙瑞才, 康俊梅, 杨青川, 李明娜. 紫花苜蓿MsCEP基因家族的鉴定及其调控根系生长发育功能的分析[J]. 中国农业科学, 2024, 57(24): 4839-4853. |
| [11] | 张荣, 刘淋茹, 付凯霞, 武紫君, 宋一凡, 王璐媛, 侯阁阁, 贺利, 冯伟, 段剑钊, 王永华, 郭天财. 干旱胁迫下外源褪黑素对冬小麦小花发育及碳营养代谢的调控[J]. 中国农业科学, 2024, 57(23): 4644-4657. |
| [12] | 钟梓春, 吴洪鑫, 张杰, 郭玉静, 何柳燕, 许小霞, 金丰良, 庞锐. 三种稻飞虱Toll受体基因家族比较分析[J]. 中国农业科学, 2024, 57(20): 4007-4021. |
| [13] | 姜亚楠, 亓方剑, 李维维, 陈巨莲, 谭晓玲. 温度升高通过小麦根际微生物加快禾谷缢管蚜和麦长管蚜的种群增长[J]. 中国农业科学, 2024, 57(20): 4045-4056. |
| [14] | 何军敏, 毛静艺, 魏晨, 任一帆, 张果平, 田可川, 刘桂芬. 基于miRNA测序数据解析苏博美利奴羊毛囊发育分子机制[J]. 中国农业科学, 2024, 57(19): 3917-3935. |
| [15] | 朱俊杰, 张鑫悦, 潘梦影, 张静雯, 郑琦, 李玉玲, 董永彬. 玉米EIN3/EIL家族基因ZmEIL9调控籽粒发育[J]. 中国农业科学, 2024, 57(18): 3522-3532. |
|
||