






中国农业科学 ›› 2022, Vol. 55 ›› Issue (18): 3484-3500.doi: 10.3864/j.issn.0578-1752.2022.18.002
李晓川( ),王朝海,周平,马维,吴瑞,宋治豪,梅艳
),王朝海,周平,马维,吴瑞,宋治豪,梅艳
收稿日期:2022-04-12
接受日期:2022-07-07
出版日期:2022-09-16
发布日期:2022-09-22
联系方式:
李晓川,Tel:13031168647;E-mail: 475383510@qq.com
基金资助:
XiaoChuan LI(),ChaoHai WANG,Ping ZHOU,Wei MA,Rui WU,ZhiHao SONG,Yan MEI
Received:2022-04-12
Accepted:2022-07-07
Published:2022-09-16
Online:2022-09-22
摘要:
【目的】进行田间晚疫病抗性评价,利用SNP分子标记分析马铃薯抗晚疫病种质的遗传多样性及分离基因组内可能影响马铃薯晚疫病抗病性状的遗传区段,为马铃薯晚疫病抗性种质创新与利用提供理论依据。【方法】通过多年多点田间鉴定对马铃薯种质资源进行晚疫病抗性评价。利用dd-RAD技术对马铃薯种质资源进行简化基因组测序并分型SNP标记。利用Admixture软件分析群体的遗传结构,GCTA软件进行主成分分析,fastTree软件构建系统发育树,stacks程序包中的populations命令计算群体遗传多样性参数,vcftools程序计算选择性消除参数,Clustal Omega程序比对氨基酸序列,MEGA6绘制氨基酸序列进化树,GEMMA 0.98.1软件进行全基因组关联分析,CMplot程序绘制QQ图和曼哈顿图。【结果】通过对马铃薯种质资源多年多点田间晚疫病抗性进行鉴定,得到101个抗晚疫病的品种(系)及21个感病品种,并对它们进行dd-RAD简化基因组测序,通过对比参考基因组,共检测到分布相对均匀的8 697 602个SNP。通过种群结构分析、主成分分析和系统进化分析,将这些种质资源进一步划分为6个群体。6个群体之内的平均核苷酸多样性值(π)为0.2055—0.2572,之间的群体分化指数(Fst)为0.156909—0.187336,说明这些种质资源存在较丰富的单核苷酸多态性。同时6个群体内期望杂合度(He)为0.187—0.2297,观测杂合度(Ho)为0.0829—0.1186,6个群体内的观测杂合度均小于期望杂合度,并且6个群体内的近交系数(Fis)范围为0.2412—0.3554,说明在选育这些种质的过程中存在近交现象。分析可能影响晚疫病抗性的马铃薯基因组遗传区段,以20 kb为窗口,5 kb为步长,在基因组相同位置,分别计算不同抗性种质间π值比值和Fst值,进行选择性消除分析,选择π值比值最小的5%及Fst值最大的10%的745个遗传区段进行分析,遗传区段中共包含507个基因,其中,有4个NBS-LRR类基因。利用群体SNP和马铃薯种质的不同晚疫病抗性表型,进行全基因组关联分析,有9个SNP与抗病性状高度相关,其周围50 kb的基因组范围内,有69个基因,其中15个基因预测参与到应激反应,12个基因预测参与清除过氧化物自由基过程。【结论】dd-RAD简化基因组测序可以在马铃薯基因组中分型获得数量较多、分布相对均匀的SNP标记。马铃薯田间抗晚疫病种质资源拥有较丰富的单核苷酸多态性,但在其选育过程中存在近交现象。选择性清除和关联分析有助于分离影响晚疫病抗病性状的遗传区段。
李晓川,王朝海,周平,马维,吴瑞,宋治豪,梅艳. 马铃薯品种(系)田间晚疫病抗性评价和全基因组遗传多样性分析[J]. 中国农业科学, 2022, 55(18): 3484-3500.
XiaoChuan LI,ChaoHai WANG,Ping ZHOU,Wei MA,Rui WU,ZhiHao SONG,Yan MEI. Deciphering of the Genetic Diversity After Field Late Blight Resistance Evaluation of Potato Breeds[J]. Scientia Agricultura Sinica, 2022, 55(18): 3484-3500.
表1
122份马铃薯种质资源和2d-RAD简化基因组测序情况"
| 序号 Serial | 品种(系) Varieties (lines) | 数据量 Total bases (bp) | 高质量序列数 HQ reads | 序列比对数 Mapped reads | 序列比对比 Mapping rate (%) | 覆盖度 Coverage (%) | 代号* Code |
|---|---|---|---|---|---|---|---|
| 1 | 云薯801 YunShu801 | 1389519882 | 9887037 | 9820720 | 99.33 | 15.86 | X14 |
| 2 | L0277-17 | 1479741228 | 10573903 | 10516208 | 99.45 | 17.45 | X67 |
| 3 | 云薯105 YunShu105 | 1430954730 | 10217553 | 10151185 | 99.35 | 17.33 | X65 |
| 4 | 会薯14号HuiShu14 | 1475603937 | 10491378 | 10412675 | 99.25 | 17.69 | X16 |
| 5 | 会薯8号HuiShu8 | 1724226138 | 12327810 | 12244428 | 99.32 | 17.89 | X59 |
| 6 | 毕薯2号BiShu2 | 1630027647 | 11604162 | 11524406 | 99.31 | 17.50 | X56 |
| 7 | 13-10-34 | 1457291493 | 10464271 | 10391818 | 99.31 | 18.53 | X19 |
| 8 | 37-47 | 1381932198 | 9859731 | 9801329 | 99.41 | 17.06 | X20 |
| 9 | Nicola | 1517566095 | 10836793 | 10759790 | 99.29 | 17.89 | V11 |
| 10 | Katahdin | 2005973730 | 14308809 | 14210061 | 99.31 | 18.08 | V13 |
| 11 | 会薯13号HuiShu13 | 1513824426 | 10822275 | 10750664 | 99.34 | 16.87 | X53 |
| 12 | 丽薯10号LiShu10 | 2074351050 | 14945235 | 14855932 | 99.40 | 18.44 | X79 |
| 13 | 云薯107 YunShu107 | 1808600760 | 12934108 | 12849969 | 99.35 | 18.82 | X62 |
| 14 | 86-4 | 1773098289 | 12852561 | 12769137 | 99.35 | 19.27 | X78 |
| 15 | S04-109 | 1553937930 | 11214590 | 11132884 | 99.27 | 17.59 | X101 |
| 16 | 宣薯5号XuanShu5 | 1522850355 | 10889989 | 10826007 | 99.41 | 18.03 | X70 |
| 17 | 11-48 | 1544878800 | 11048219 | 10967798 | 99.27 | 17.90 | X7 |
| 18 | 定薯5号DingShu5 | 1235218653 | 8850250 | 8781613 | 99.22 | 16.64 | X33 |
| 19 | 毕薯8号BiShu8 | 1849000239 | 13074100 | 12968752 | 99.19 | 17.76 | X36 |
| 20 | 288-39 | 2031266196 | 14592056 | 14486152 | 99.27 | 18.60 | X41 |
| 21 | 92-101 | 1427750136 | 10224526 | 10132842 | 99.10 | 16.06 | X31 |
| 22 | 167-327 | 1233568926 | 8833599 | 8752753 | 99.08 | 15.32 | X24 |
| 23 | 77-29 | 1607038884 | 11485513 | 11406544 | 99.31 | 18.07 | X8 |
| 24 | Agria | 1873257894 | 13391597 | 13304811 | 99.35 | 18.36 | V4 |
| 25 | 定薯3号DingShu3 | 1939569498 | 14050420 | 13959591 | 99.35 | 21.62 | X95 |
| 26 | 鄂薯18号EShu18 | 1748400651 | 12591486 | 12507551 | 99.33 | 19.14 | X94 |
| 27 | A153 | 1546019631 | 11036891 | 10967645 | 99.37 | 17.67 | X58 |
| 28 | 77-21 | 2340564876 | 16498728 | 16352763 | 99.12 | 27.14 | X4 |
| 29 | 43-36 | 1785437622 | 12769189 | 12689774 | 99.38 | 18.40 | X38 |
| 30 | 104-376 | 2081927016 | 15013206 | 14909294 | 99.31 | 19.49 | X75 |
| 31 | 198-151 | 1506050649 | 10766415 | 10707633 | 99.45 | 18.30 | X69 |
| 32 | 225-177 | 1428782715 | 10123094 | 10060117 | 99.38 | 17.33 | X12 |
| 33 | 43-20 | 1217961666 | 8719306 | 8665729 | 99.39 | 16.04 | X32 |
| 34 | 中薯17号ZhongShu17 | 1636463898 | 11773432 | 11589241 | 98.44 | 20.53 | V21 |
| 35 | Favorita | 1749736224 | 12528593 | 12449651 | 99.37 | 19.32 | V3 |
| 36 | Russet Burbank | 1915560153 | 13773752 | 13690282 | 99.39 | 22.08 | V20 |
| 37 | Norchip | 1232854965 | 8755118 | 8690255 | 99.26 | 16.97 | V16 |
| 38 | Spunta | 1701510795 | 12281997 | 12189765 | 99.25 | 17.37 | V1 |
| 39 | S10-557 | 1800904824 | 12904151 | 12826113 | 99.40 | 16.87 | X74 |
| 40 | S10-676 | 1875586428 | 13457582 | 13376099 | 99.39 | 17.68 | X71 |
| 41 | S04-921 | 1369934361 | 9804477 | 9747198 | 99.42 | 17.17 | X66 |
| 42 | 云薯902 YunShu902 | 2269214694 | 16348796 | 16240617 | 99.34 | 18.44 | X89 |
| 43 | LZ111 | 1701482616 | 12279442 | 12210831 | 99.44 | 18.88 | X77 |
| 44 | S06-277 | 1976278086 | 14233248 | 14130408 | 99.28 | 18.36 | X100 |
| 45 | A89 | 1705085901 | 12127056 | 12058833 | 99.44 | 19.33 | X68 |
| 46 | Kennebec | 1894451013 | 13515960 | 13429500 | 99.36 | 18.93 | V2 |
| 47 | 12-9 | 1642531869 | 11661138 | 11584044 | 99.34 | 16.91 | X55 |
| 48 | 云薯1号YunShu1 | 1932156747 | 13982565 | 13887872 | 99.32 | 20.44 | X96 |
| 49 | 云薯505 YunShu505 | 2278091020 | 16058092 | 15921163 | 99.15 | 25.27 | X3 |
| 50 | 丽薯13号LiShu13 | 1792673208 | 12943700 | 12860661 | 99.36 | 18.37 | X90 |
| 51 | 宣薯6号XuanShu6 | 1743643143 | 12416240 | 12333510 | 99.33 | 18.19 | X57 |
| 52 | 16-811-92 | 2571656553 | 18433648 | 18323584 | 99.40 | 18.79 | X81 |
| 53 | 16-81-19 C5 | 1566249921 | 11152065 | 11073521 | 99.30 | 18.36 | X44 |
| 54 | 105-148 | 1830419676 | 13169922 | 13060177 | 99.17 | 18.40 | X72 |
| 55 | YS481 | 2176126065 | 15681885 | 15582664 | 99.37 | 18.08 | X92 |
| 56 | Superior | 1795602987 | 12872912 | 12799481 | 99.43 | 19.35 | V12 |
| 57 | 毕薯10号BiShu10 | 1946519667 | 13906796 | 13820590 | 99.38 | 18.21 | X40 |
| 58 | 10-61 | 1881900756 | 13451123 | 13366356 | 99.37 | 18.91 | X64 |
| 59 | 32-80 | 1839399849 | 13188933 | 13101780 | 99.34 | 19.30 | X35 |
| 60 | 19-182 | 1542493908 | 10949872 | 10868390 | 99.26 | 18.30 | X9 |
| 61 | 19-75-22 | 1418241816 | 10277631 | 10215203 | 99.39 | 20.37 | X48 |
| 62 | 33-46 | 1433188962 | 10193577 | 10125468 | 99.33 | 16.44 | X15 |
| 63 | C149 | 1663146900 | 11887115 | 11796792 | 99.24 | 18.77 | X6 |
| 64 | 6-25 | 1966347081 | 14091871 | 13993199 | 99.30 | 19.31 | X39 |
| 65 | Desiree | 1741237884 | 12436565 | 12357551 | 99.36 | 18.31 | V5 |
| 66 | Hertha | 1184096367 | 8370420 | 8327303 | 99.48 | 16.57 | V15 |
| 67 | 4-151 | 1457695764 | 10419341 | 10333207 | 99.17 | 17.23 | X5 |
| 68 | 25-2 | 1341393498 | 9611717 | 9544222 | 99.30 | 16.70 | X34 |
| 69 | 24-37-55 | 1976929830 | 14344412 | 14259611 | 99.41 | 21.19 | X86 |
| 70 | Chieftain | 1999068759 | 14306469 | 14194123 | 99.21 | 19.47 | V6 |
| 71 | Shepody | 1696532877 | 12146034 | 12060607 | 99.30 | 18.70 | V9 |
| 72 | Ranger Russet | 1745461386 | 12518041 | 12422160 | 99.23 | 19.28 | V18 |
| 73 | Snowden | 2128781160 | 15119156 | 15004489 | 99.24 | 19.65 | V7 |
| 74 | Felsina | 2037316590 | 14511003 | 14421521 | 99.38 | 21.57 | V19 |
| 75 | Atlantic | 1752787368 | 12542637 | 12455169 | 99.30 | 19.79 | V8 |
| 76 | Lenape | 1054845711 | 7540802 | 7470305 | 99.07 | 17.88 | V17 |
| 77 | 丽薯18号LiShu18 | 1983366639 | 14258000 | 14169149 | 99.38 | 19.30 | X73 |
| 78 | BP0806-30 | 1665025128 | 11779252 | 11694069 | 99.28 | 18.13 | X60 |
| 79 | BP0806-21 | 1712587932 | 12322410 | 12234910 | 99.29 | 17.99 | X106 |
| 80 | 鄂薯14号EShu14 | 957120660 | 6750190 | 6701577 | 99.28 | 16.68 | X2 |
| 81 | Belchip | 994979286 | 7074600 | 6985059 | 98.73 | 16.27 | V14 |
| 82 | Innovator | 2129532228 | 15201441 | 15094352 | 99.30 | 20.18 | V10 |
| 83 | 24-2-74 | 1512989658 | 10836722 | 10732355 | 99.04 | 17.65 | X47 |
| 84 | 丽薯6号LiShu6 | 1335983409 | 9559009 | 9479823 | 99.17 | 18.40 | X29 |
| 85 | NSN8 | 1707356961 | 12237252 | 12148835 | 99.28 | 17.14 | X98 |
| 86 | YSN3 | 1918037115 | 13780834 | 13684547 | 99.30 | 18.49 | X93 |
| 87 | 合作88 HeZuo88 | 1241992494 | 8892503 | 8826214 | 99.25 | 17.50 | X49 |
| 88 | cip88 | 1592733159 | 11381458 | 11294212 | 99.23 | 18.37 | X50 |
| 89 | c88 | 1114612254 | 7962710 | 7903628 | 99.26 | 16.30 | X51 |
| 90 | 威芋6号WeiYu6 | 1698505965 | 12078111 | 11949277 | 98.93 | 17.52 | X17 |
| 91 | 威芋7号WeiYu7 | 1089760050 | 7749041 | 7680101 | 99.11 | 15.71 | X1 |
| 92 | V2011-2 | 1350470763 | 9632054 | 9534464 | 98.99 | 17.40 | X11 |
| 93 | Z59 | 1266584391 | 9067253 | 9018186 | 99.46 | 18.61 | X63 |
| 94 | 12-1-4 | 1493397441 | 10657657 | 10595044 | 99.41 | 18.36 | X61 |
| 95 | WY08002 | 1340769375 | 9582631 | 9512250 | 99.27 | 16.92 | X52 |
| 96 | 11-86-90 | 2101533741 | 15105304 | 15001569 | 99.31 | 18.42 | X83 |
| 97 | 青薯9号QingShu9 | 1995336018 | 14349098 | 14254442 | 99.34 | 18.41 | X76 |
| 98 | 云薯401 YunShu401 | 1565585901 | 11348147 | 11277951 | 99.38 | 20.10 | X42 |
| 99 | 会薯15号HuiShu15 | 1314918630 | 9407429 | 9347588 | 99.36 | 16.58 | X30 |
| 100 | 0401-11 | 1672961841 | 12084421 | 11968695 | 99.04 | 19.40 | X99 |
| 101 | 威薯9号WeiShu9 | 1728903294 | 12517795 | 12432832 | 99.32 | 19.66 | X97 |
| 102 | H3 | 1367316783 | 9812205 | 9723683 | 99.10 | 17.92 | X54 |
| 103 | 2005-1 | 1671491790 | 12050460 | 11974544 | 99.37 | 17.86 | X104 |
| 104 | 23-61-259 | 1225347075 | 8768959 | 8702442 | 99.24 | 15.53 | X25 |
| 105 | 19-61-96 | 2114742438 | 15292497 | 15184121 | 99.29 | 20.07 | X87 |
| 106 | 27-81-72 | 1291962231 | 9201567 | 9126131 | 99.18 | 17.03 | X28 |
| 107 | 会薯16号HuiShu16 | 1872061542 | 13363101 | 13282357 | 99.40 | 19.22 | X37 |
| 108 | 川芋A50 ChuanYuA50 | 1426170996 | 10109290 | 10017617 | 99.09 | 17.12 | X13 |
| 109 | 23-13-5 | 1264771449 | 9051622 | 8975710 | 99.16 | 16.74 | X27 |
| 110 | 23-57-63 | 1244052630 | 8919865 | 8861964 | 99.35 | 16.49 | X26 |
| 111 | 黔芋7号QianYu7 | 1345688424 | 9563048 | 9495424 | 99.29 | 18.20 | X10 |
| 112 | 22-91-24 | 1732358151 | 12397229 | 12322974 | 99.40 | 18.39 | X85 |
| 113 | 18-81-325 | 1247259735 | 8913644 | 8805094 | 98.78 | 16.25 | X21 |
| 114 | 22-92-256 | 1175470803 | 8422225 | 8337731 | 99.00 | 16.58 | X22 |
| 115 | 18-91-127 | 1660783212 | 11832737 | 11752580 | 99.32 | 18.56 | X45 |
| 116 | 27-3-52 | 2069760663 | 14913871 | 14796774 | 99.21 | 18.70 | X82 |
| 117 | 200-10-50 | 1721414934 | 12260394 | 12144291 | 99.05 | 19.31 | X18 |
| 118 | 225-10-21 | 1145114208 | 8192235 | 8107691 | 98.97 | 15.94 | X23 |
| 119 | 22-83-38 | 1928771919 | 13864980 | 13742071 | 99.11 | 18.21 | X88 |
| 120 | 278-10-74 | 2072509092 | 14889774 | 14797496 | 99.38 | 18.15 | X80 |
| 121 | 24-79-59 | 1460479347 | 10417428 | 10343662 | 99.29 | 17.79 | X46 |
| 122 | 22-69-90 | 1375713009 | 9810708 | 9739261 | 99.27 | 17.58 | X43 |
表2
122份马铃薯种质资源SNP基因分型情况"
| SNP类型SNP type | 数目Number | 占比Percentage (%) | |
|---|---|---|---|
| 外显子区域Exotic total | 同义突变Synonymous SNP | 182507 | 2.10 |
| 非同义突变Nonsynonymous SNP | 209566 | 2.41 | |
| 获得终止密码子的 SNP Stopgain | 4602 | 0.05 | |
| 丢失终止密码子的 SNP Stoploss | 569 | 0.01 | |
| 总计Total | 397244 | 4.57 | |
| 拼接接头区域Splicing | 1762 | 0.02 | |
| 内含子区域Intronic | 970743 | 11.16 | |
| 基因间区域Intergenic | 6650230 | 76.46 | |
| 5′UTR区域5′UTR | 46263 | 0.53 | |
| 3′UTR区域3′UTR | 98348 | 1.13 | |
| UTR5; UTR3 | 29 | 0.00 | |
| 转录起始位点上游1 kb的区域Upstream 1 kb | 247351 | 2.84 | |
| 转录终止位点下游1 kb的区域Downstream 1 kb | 251705 | 2.89 | |
| 其他Others | 33956 | 0.39 | |
| 总计Total | 8697602 | 100 | |
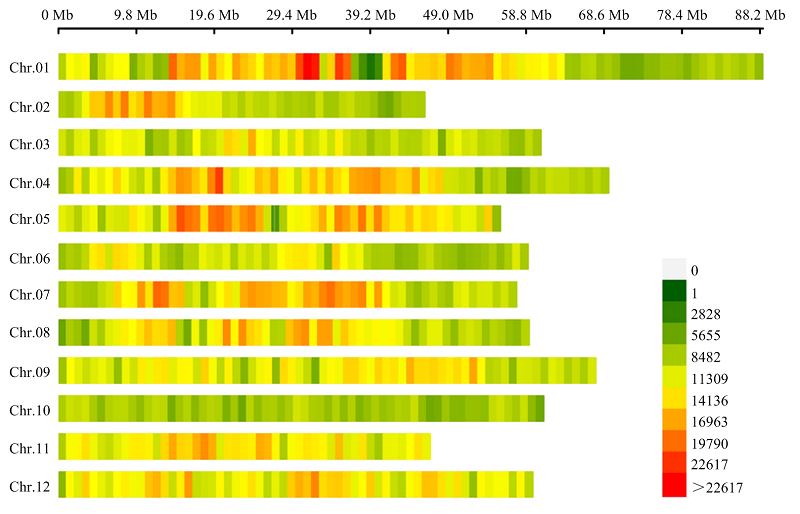
图1
2d-RAD简化基因组测序分型得到的SNP密度热图热图以1 Mb的窗口大小显示,图右侧为各颜色表示的SNP数量"

图2
122份马铃薯种质资源的种群分析 A:当K=2—20时,种群结构分析的cv error值;B:当K=2—20时,122份马铃薯种质资源的种群结构;C:122份马铃薯种质资源的主成分分析;D:122份马铃薯种质资源的系统进化树"
表3
群体内的遗传多样性参数"
| 群体 Population | 核苷酸多样性 Pi (π) | 观测杂合度 Obs Het (Ho) | 期望杂合度 Exp Het (He) | 近交系数 Fis |
|---|---|---|---|---|
| Ⅰ | 0.2055 | 0.0829 | 0.187 | 0.2858 |
| Ⅱ | 0.2572 | 0.1186 | 0.2297 | 0.3319 |
| Ⅲ | 0.2137 | 0.091 | 0.1913 | 0.2716 |
| Ⅳ | 0.2188 | 0.1051 | 0.1942 | 0.2412 |
| Ⅴ | 0.2098 | 0.0918 | 0.1898 | 0.2692 |
| Ⅵ | 0.2319 | 0.0838 | 0.2124 | 0.3554 |
表4
群体间群体分化指数(Fst)的矩阵"
| 群体 Population | II | III | IV | V | VI |
|---|---|---|---|---|---|
| I | 0.159407 | 0.172889 | 0.172479 | 0.174660 | 0.165741 |
| II | 0.163408 | 0.156909 | 0.176573 | 0.170987 | |
| III | 0.170947 | 0.183461 | 0.158484 | ||
| IV | 0.187336 | 0.159748 | |||
| V | 0.160706 |
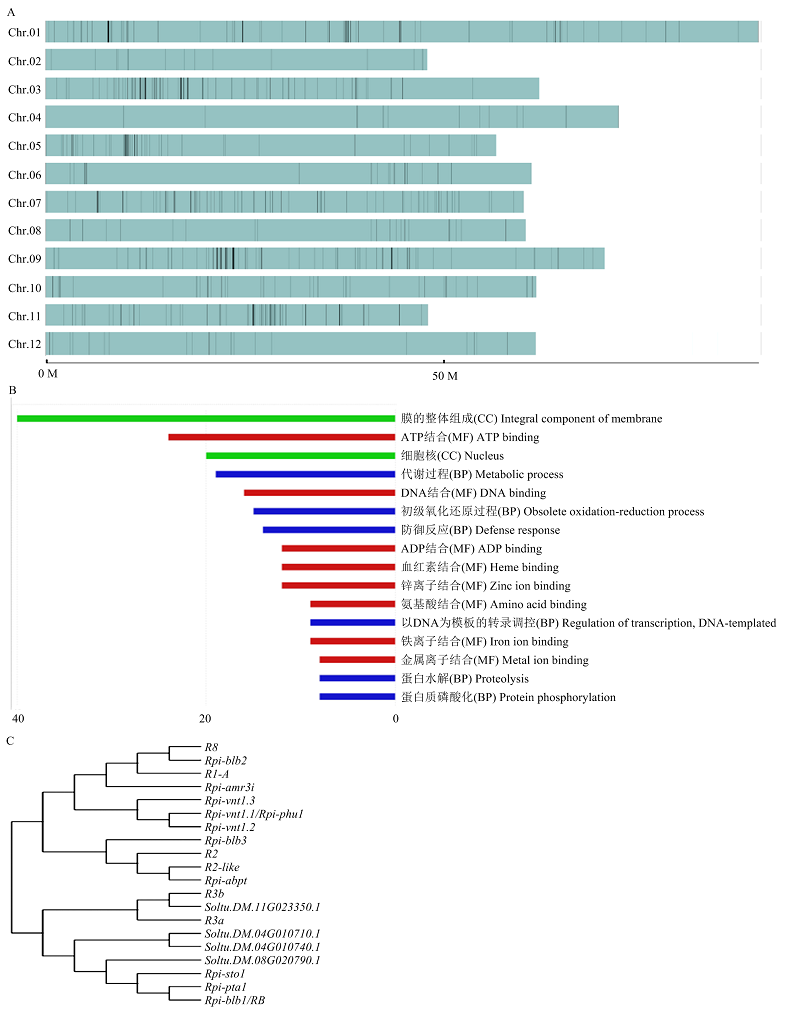
图3
选择性清除分析 A:745个可能影响马铃薯晚疫病抗病性状的受选择遗传区段在染色体上的分布图;B:受选择遗传区段中,基因数量较多的几个GO条目及基因数量;C:4个NBS-LRR类基因与已克隆晚疫病抗病(R)基因的遗传进化树"
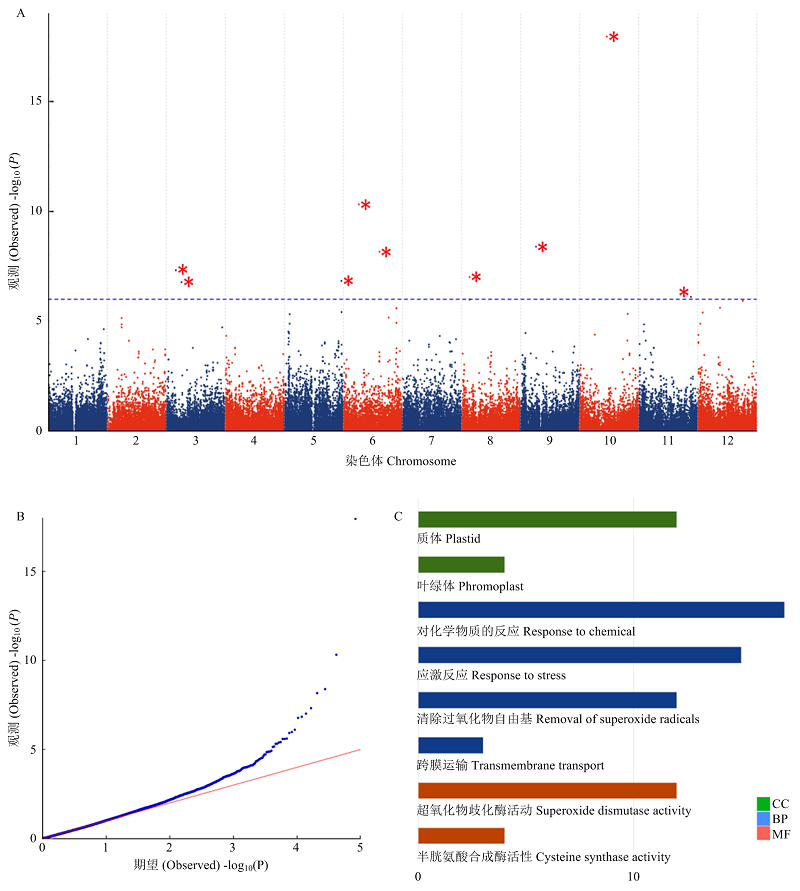
图4
全基因组关联分析 A:各SNP位点的-log10(P)在马铃薯基因组中分布的曼哈顿图;B:全基因组关联分析中各SNP位点的期望-log10(P)和观测-log10(P)的QQ图;C:与晚疫病抗性相关的SNP位点附近的基因组中,基因数量较多的几个GO条目"
| [1] | KAMOUN S. Nonhost resistance to Phytophthora: Novel prospects for a classical problem. Current Opinion in Plant Biology, 2001, l4: 295-300. |
| [2] |
GUO L, ZHU X Q, HU C H, RISTAINO J B. Genetic structure of Phytophthora infestans populations in China indicates multiple migration events. Phytopathology, 2010, 100(10): 997.
doi: 10.1094/PHYTO-05-09-0126 |
| [3] | 杨露, 王勇, 吴石平. 贵州马铃薯晚疫病菌群体遗传多样性分析. 贵州农业科学, 2019, 47(3): 64-67. |
| YANG L, WANG Y, WU S P. Genetic diversity analysis of Phytophthora infestans population in Guizhou province. Guizhou Agriculture Science, 2019, 47(3): 64-67. (in Chinese) | |
| [4] |
YUEN J E, ANDERSSON B. What is the evidence for sexual reproduction of Phytophthora infestans in Europe? Plant Pathology, 2013, 62(3): 485-491.
doi: 10.1111/j.1365-3059.2012.02685.x |
| [5] |
VAN DER LEE T, TESTA A, VAN'T KLOOSTER J, VAN DEN BERG-VELTHUIS G, GOVERS F. Chromosomal deletion in isolates of Phytophthora infestans correlates with virulence on R3, R10, and R11 potato lines. Molecular Plant-Microbe Interactions, 2001, 14(12): 1444-1452.
doi: 10.1094/MPMI.2001.14.12.1444 |
| [6] |
PARK T H, VLEESHOUWERS V G A A, JACOBSEN E, VAN DER VOSSEN E, VISSER R G F. Molecular breeding for resistance to Phytophthora infestans (Mont.) de Bary in potato (Solanum tuberosum L.): A perspective of cisgenesis. Plant Breeding, 2009, 128(2): 109-117.
doi: 10.1111/j.1439-0523.2008.01619.x |
| [7] |
VAN DER VOSSEN E A, GROS J, SIKKEMA A, MUSKENS M, WOUTERS D, WOLTERS P, PEREIRA A, ALLEFS S. The Rpi-blb2 gene from Solanum bulbocastanum is an Mi-1 gene homolog conferring broad-spectrum late blight resistance in potato. The Plant Journal, 2005, 44(2): 208-222.
doi: 10.1111/j.1365-313X.2005.02527.x |
| [8] |
BALLVORA A, ERCOLANO M R, WEISS J, MEKSEM K, BORMANN C A, OBERHAGEMANN P, SALAMINI F, GEBHARDT C. The R1 gene for potato resistance to late blight (Phytophthora infestans) belongs to the leucine zipper/NBS/LRR class of plant resistance genes. The Plant Journal, 2002, 30(3): 361-371.
doi: 10.1046/j.1365-313X.2001.01292.x |
| [9] |
LOKOSSOU A A, PARK T H, VAN ARKEL G, ARENS M, RUYTER-SPIRA C, MORALES J, WHISSON S C, BIRCH P R, VISSER R G, JACOBSEN E, VAN DER VOSSEN E A. Exploiting knowledge of R/Avr genes to rapidly clone a new LZ-NBS-LRR family of late blight resistance genes from potato linkage group IV. Molecular Plant-Microbe Interactions, 2009, 22(6): 630-641.
doi: 10.1094/MPMI-22-6-0630 |
| [10] |
HUANG S, VAN DER VOSSEN E A, KUANG H, VLEESHOUWERS V G, ZHANG N, BORM T J, VAN ECK H J, BAKER B, JACOBSEN E, VISSER R G. Comparative genomics enabled the isolation of the R3a late blight resistance gene in potato. The Plant Journal, 2005, 42: 251-261.
doi: 10.1111/j.1365-313X.2005.02365.x |
| [11] |
LI G, HUANG S, GUO X, LI Y, YANG Y, GUO Z, KUANG H, RIETMAN H, BERGERVOET M, VLEESHOUWERS V G, VAN DER VOSSEN E A, QU D, VISSER R G, JACOBSEN E, VOSSEN J H. Cloning and characterization of R3b; Members of the R3 superfamily of late blight resistance genes show sequence and functional divergence. Molecular Plant-Microbe Interactions, 2011, 24(10): 1132-1142.
doi: 10.1094/MPMI-11-10-0276 |
| [12] | VOSSEN J H, VAN ARKEL G, BERGERVOET M, JO K R, JACOBSEN E, VISSER R G. The Solanum demissum R8 late blight resistance gene is an Sw-5 homologue that has been deployed worldwide in late blight resistant varieties. Theoretical and Applied Genetics, 2016, 129(9): 1785-1796. |
| [13] |
VAN DER VOSSEN E, SIKKEMA A, HEKKERT B T, GROS J, STEVENS P, MUSKENS M, WOUTERS D, PEREIRA A, STIEKEMA W, ALLEFS S. An ancient R gene from the wild potato species Solanum bulbocastanum confers broad-spectrum resistance to Phytophthora infestans in cultivated potato and tomato. The Plant Journal, 2003, 36: 867-882.
doi: 10.1046/j.1365-313X.2003.01934.x |
| [14] | SONG J, BRADEEN J M, NAESS S K, RAASCH J A, WIELGUS S M, HABERLACH G T, LIU J, KUANG H, AUSTIN-PHILLIPS S, BUELL C R, HELGESON J P, JIANG J. Gene RB cloned from Solanum bulbocastanum confers broad spectrum resistance to potato late blight. Proceedings of the National Academy of Sciences of the United States of America, 2003, 100(16): 9128-9133. |
| [15] |
VLEESHOUWERS V G, RIETMAN H, KRENEK P, CHAMPOURET N, YOUNG C, OH S K, WANG M, BOUWMEESTER K, VOSMAN B, VISSER R G, JACOBSEN E, GOVERS F, KAMOUN S, VAN DER VOSSEN E A. Effector genomics accelerates discovery and functional profiling of potato disease resistance and Phytophthora infestans avirulence genes. PLoS ONE, 2008, 3: e2875.
doi: 10.1371/journal.pone.0002875 |
| [16] |
WANG M, ALLEFS S, VAN DEN BERG R G, VLEESHOUWERS V G, VAN DER VOSSEN E A, VOSMAN B. Allele mining in Solanum: Conserved homologues of Rpi-blb1 are identified in Solanum stoloniferum. Theoretical and Applied Genetics, 2008, 116(7): 933-943.
doi: 10.1007/s00122-008-0725-3 |
| [17] |
FOSTER S J, PARK T H, PEL M, BRIGNETI G, SLIWKA J, JAGGER L, VAN DER VOSSEN E, JONES J D. Rpi-vnt1.1, a Tm-22 homolog from Solanum venturii, confers resistance to potato late blight. Molecular Plant- Microbe Interactions, 2009, 22: 589-600.
doi: 10.1094/MPMI-22-5-0589 |
| [18] |
ŚLIWKA J, ŚWIĄTEK M, TOMCZYŃSKA I, STEFAŃCZYK E, CHMIELARZ M, ZIMNOCH- GUZOWSKA E. Influence of genetic background and plant age on expression of the potato late blight resistance gene Rpi-phu1during incompatible interactions with Phytophthora infestans. Plant Pathology, 2013, 62(5): 1072-1080.
doi: 10.1111/ppa.12018 |
| [19] |
WITEK K, JUPE F, WITEK A I, BAKER D, CLARK M D, JONES J D. Accelerated cloning of a potato late blight-resistance gene using Ren Seq and SMRT sequencing. Nature Biotechnology, 2016, 34: 656-660.
doi: 10.1038/nbt.3540 |
| [20] |
HERMSEN J G T H, RAMANNA M S. Double-bridge hybrids of Solanum bulbocastanum and cultivars of Solanum tuberosum. Euphytica, 1973, 22(3): 457-466.
doi: 10.1007/BF00036641 |
| [21] |
HAVERKORT A J, BOONEKAMP P M, HUTTEN R, JACOBSEN E, LOTZ L A P, KESSELG J T, VOSSEN J H, VISSER R G F. Durable late blight resistance in potato through dynamic varieties obtained by cisgenesis: Scientific and societal advances in the DuRPh project. Potato Research, 2016, 59(1): 35-66.
doi: 10.1007/s11540-015-9312-6 |
| [22] |
PETERSON B K, WEBER J N, KAY E H, FISHER H S, HOEKSTRA H E. Double digest RADseq: An inexpensive method for De Novo SNP discovery and genotyping in model and non-model species. PLoS ONE, 2012, 7(5): e37135.
doi: 10.1371/journal.pone.0037135 |
| [23] | SEVERN-ELLIS A A, SCHEBEN A, NEIK TX, SAAD NSM, PRADHAN A, BATLEY J. Genotyping for species identification and diversity assessment using double-digest restriction site-associated DNA sequencing (ddRAD-Seq). Methods in Molecular Biology, 2020, 2107: 159-187. |
| [24] | CHEN S, ZHOU Y, CHEN Y, GU J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 2018, 34(17): i884-i890. |
| [25] |
LI H, DURBIN R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009, 25(14): 1754-1760.
doi: 10.1093/bioinformatics/btp324 |
| [26] |
POTATO GENOME SEQUENCING CONSORTIUM. Genome sequence and analysis of the tuber crop potato. Nature, 2011, 475: 189-195.
doi: 10.1038/nature10158 |
| [27] |
PHAM G M, HAMILTON J P, WOOD J C, BURKE J T, ZHAO H, VAILLANCOURT B, OU S, JIANG J, BUELL C R. Construction of a chromosome-scale long-read reference genome assembly for potato. GigaScience, 2020, 9(9): giaa100.
doi: 10.1093/gigascience/giaa100 |
| [28] |
ZHU P, HE L, LI Y, HUANG W, XI F, LIN L, ZHI Q, ZHANG W, TANG Y T, GENG C, LU Z, XU X. OTG- snpcaller: An optimized pipeline based on TMAP and GATK for SNP calling from ion torrent data. PLoS ONE, 2014, 9(5): e97507.
doi: 10.1371/journal.pone.0097507 |
| [29] |
WANG K, LI M, HAKONARSON H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research, 2010, 38(16): e164.
doi: 10.1093/nar/gkq603 |
| [30] |
YANG J, LEE S H, GODDARD M E, VISSCHER P M. GCTA: A tool for genome-wide complex trait analysis. American Journal of Human Genetics, 2011, 88(1): 76-82.
doi: 10.1016/j.ajhg.2010.11.011 |
| [31] |
PRICE M N, DEHAL P S, ARKIN A P. FastTree 2--Approximately maximum-likelihood trees for large alignments. PLoS ONE, 2010, 5(3): e9490.
doi: 10.1371/journal.pone.0009490 |
| [32] |
ALEXANDER D H, NOVEMBRE J, LANGE K. Fast model-based estimation of ancestry in unrelated individuals. Genome Research, 2009, 19: 1655-1664.
doi: 10.1101/gr.094052.109 |
| [33] |
DAVEY J W, HOHENLOHE P A, ETTER P D, BOONE J Q, CATCHEN J M, BLAXTER M L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics, 2011, 12(7): 499-510.
doi: 10.1038/nrg3012 |
| [34] |
DANECEK P, AUTON A, ABECASIS G, ALBERS C A, BANKS E, DEPRISTO M A, HANDSAKER R E, LUNTER G, MARTH G T, SHERRY S T, MCVEAN G, DURBIN R, 1000 GENOMES PROJECT ANALYSIS GROUP. The variant call format and VCFtools. Bioinformatics, 2011, 27(15): 2156-2158.
doi: 10.1093/bioinformatics/btr330 |
| [35] | RAUDVERE U, KOLBERG L, KUZMIN I, ARAK T, ADLER P, PETERSON H, VILO J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists. Nucleic Acids Research, 2019, 47(W1): 191-198. |
| [36] |
SIEVERS F, WILM A, DINEEN D, GIBSON T J, KARPLUS K, LI W, LOPEZ R, MCWILLIAM H, REMMERT M, SÖDING J, THOMPSON J D, HIGGINS D G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology, 2011, 7: 539.
doi: 10.1038/msb.2011.75 |
| [37] |
TAMURA K, STECHER G, PETERSON D, FILIPSKI A, KUMAR S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular Biology and Evolution, 2013, 30: 2725-2729.
doi: 10.1093/molbev/mst197 |
| [38] |
ZHOU X, STEPHENS M. Genome-wide efficient mixed-model analysis for association studies. Nature Genetics, 2012, 44(7): 821-824.
doi: 10.1038/ng.2310 |
| [39] |
UITDEWILLIGEN J G, WOLTERS A M, D’HOOP B B, BORM T J, VISSER R G, VAN ECK H J. A next- generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS ONE, 2013, 8: e62355.
doi: 10.1371/journal.pone.0062355 |
| [40] |
WANG S, MEYER E, MCKAY J K, MATZ M V. 2b-rad: A simple and flexible method for genome-wide genotyping. Nature Methods, 2012, 9: 808-810.
doi: 10.1038/nmeth.2023 |
| [41] |
VOS P G, PAULO M J, VOORRIPS R E, VISSER R G, VAN ECK H J, VAN EEUWIJK F A. Evaluation of LD decay and various LD-decay estimators in simulated and SNP-array data of tetraploid potato. Theoretical and Applied Genetics, 2017, 130: 123-135.
doi: 10.1007/s00122-016-2798-8 |
| [42] |
D'HOOP B B, PAULO M J, KOWITWANICH K, SENGERS M, VISSER R G, VAN ECK H J, VAN EEUWIJK F A. Population structure and linkage disequilibrium unravelled in tetraploid potato. Theoretical and Applied Genetics, 2010, 121: 1151-1170.
doi: 10.1007/s00122-010-1379-5 |
| [43] |
SIMKO I, COSTANZO S, HAYNES K G, CHRIST B J, JONES R W. Linkage disequilibrium mapping of a Verticillium dahliae resistance quantitative trait locus in tetraploid potato (Solanum tuberosum) through a candidate gene approach. Theoretical and Applied Genetics, 2004, 108: 217-224.
doi: 10.1007/s00122-003-1431-9 |
| [44] |
DUAN Y, DUAN S, XU J, ZHENG J, HU J, LI X, LI B, LI G, JIN L. Late blight resistance evaluation and genome- wide assessment of genetic diversity in wild and cultivated potato species. Frontiers in Plant Science, 2021, 12: 710468.
doi: 10.3389/fpls.2021.710468 |
| [45] |
WANG Y, RASHID MAR, LI X, YAO C, LU L, BAI J, LI Y, XU N, YANG Q, ZHANG L, BRYAN GJ, SUI Q PAN Z. Collection and evaluation of genetic diversity and population structure of potato landraces and varieties in China. Frontiers in Plant Science, 2019, 10: 139.
doi: 10.3389/fpls.2019.00139 |
| [46] | HIRSCH C N, HIRSCH C D, FELCHER K, COOMBS J, ZARKA D, VAN DEYNZE A, DE JONG W, VEILLEUX R E, JANSKY S, BETHKE P, DOUCHES D S, BUELL C R. Retrospective view of North American potato (Solanum tuberosum L.) breeding in the 20th and 21st centuries. G3-Genes Genomes Genetics, 2013, 3(6): 1003-1013. |
| [47] |
ABOU-TALEB E M, ABOSHOSHA S M, EL-SHERIF E M, EL-KOMY M H. Genetic diversity among late blight resistant and susceptible potato genotypes. Saudi Journal of Biological Sciences, 2010, 17(2): 133-138.
doi: 10.1016/j.sjbs.2010.02.006 |
| [48] |
HUANG X H, HAN B. Natural variations and genome-wide association studies in crop plants. Annual Review of Plant Biology, 2014, 65: 531-551.
doi: 10.1146/annurev-arplant-050213-035715 |
| [49] |
LI X, XU J, DUAN S, BIAN C, HU J, SHEN H, LI G, JIN L. Pedigree-based deciphering of genome-wide conserved patterns in an elite potato parental line. Frontiers in Plant Science, 2018, 9: 690.
doi: 10.3389/fpls.2018.00690 |
| [50] |
DUAN Y, LIU J, BIAN C, DUAN S, XU J, JIN, L. Construction of fingerprinting and analysis of genetic diversity with SSR markers for eighty-eight approved potato cultivars (Solanum tuberosum L.) in China. Acta Agronomica Sinica, 2009, 35: 1451-1457.
doi: 10.3724/SP.J.1006.2009.01451 |
| [51] |
DUAN Y, LIU J, XU J, BIAN C, DUAN S, PANG W, HU J, LI G, JIN L. DNA fingerprinting and genetic diversity analysis with simple sequence repeat markers of 217 potato cultivars (Solanum tuberosum L.) in China. American Journal of Potato Research, 2018, 96: 21-32.
doi: 10.1007/s12230-018-9685-6 |
| [52] | PLAISTED R, HOOPES R. The past record and future prospects for the use of exotic potato germplasm. American Journal of Potato Research, 1989, 66: 603-627. |
| [53] | 隋启君. 中国马铃薯育种对策浅见. 中国马铃薯, 2001, 15(5): 259-264. |
| SUI Q J. Some suggestions of improving the work if potato breeding in China. China Potato, 2001, 15(5): 259-264. (in Chinese) | |
| [54] | NEI M. Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the United States of America, 1973, 70(12): 3321-3323. |
| [55] |
NEI M. F-statistics and analysis of gene diversity in subdivided populations. Annals of Human Genetics, 1977, 41(2): 225-233.
doi: 10.1111/j.1469-1809.1977.tb01918.x |
| [56] |
ZHOU Q, TANG D, HUANG W, YANG Z, ZHANG Y, HAMILTON J P, VISSER R, BACHEM C, BUELL C R, ZHANG Z, ZHANG C, HUANG S. Haplotype-resolved genome analyses of a heterozygous diploid potato. Nature Genetics, 2020, 52(10): 1018-1023.
doi: 10.1038/s41588-020-0699-x |
| [57] |
NEI M, MILLER J C. A simple method for estimating average number of nucleotide substitutions within and between populations from restriction data. Genetics, 1990, 125(4): 873-879.
doi: 10.1093/genetics/125.4.873 |
| [58] |
HOLSINGER K E, WEIR B S. Genetics in geographically structured populations: Defining, estimating and interpreting F(ST). Nature Reviews Genetics, 2009, 10(9): 639-650.
doi: 10.1038/nrg2611 |
| [59] |
NIELSEN R, WILLIAMSON S, KIM Y, HUBISZ M J, CLARK A G, BUSTAMANTE C. Genomic scans for selective sweeps using SNP data. Genome Research, 2005, 15(11): 1566-1575.
doi: 10.1101/gr.4252305 |
| [60] |
ZENG L, TU X L, DAI H, HAN F M, LU B S, WANG M S, NANAEI H A, TAJABADIPOUR A, MANSOURI M, LI X L, JI L L, IRWIN D M, ZHOU H, LIU M, ZHENG H K, ESMAILIZADEH A, WU D D. Whole genomes and transcriptomes reveal adaptation and domestication of pistachio. Genome Biology, 2019, 20(1): 79.
doi: 10.1186/s13059-019-1686-3 |
| [61] |
LU K, WEI L, LI X, WANG Y, WU J, LIU M, ZHANG C, CHEN Z, XIAO Z, JIAN H, CHENG F, ZHANG K, DU H, CHENG X, QU C, QIAN W, LIU L, WANG R, ZOU Q, YING J, XU X, MEI J, LIANG Y, CHAI YR, TANG Z, WAN H, NI Y, HE Y, LIN N, FAN Y, SUN W, LI N N, ZHOU G, ZHENG H, WANG X, PATERSON A H, LI J. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nature Communications, 2019, 10(1): 1154.
doi: 10.1038/s41467-019-09134-9 |
| [62] |
WU D, LIANG Z, YAN T, XU Y, XUAN L, TANG J, ZHOU G, LOHWASSER U, HUA S, WANG H, CHEN X, WANG Q, ZHU L, MAODZEKA A, HUSSAIN N, LI Z, LI X, SHAMSI IH, JILANI G, WU L, ZHENG H, ZHANG G, CHALHOUB B, SHEN L, YU H, JIANG L. Whole-genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Molecular Plant, 2019, 12(1): 30-43.
doi: 10.1016/j.molp.2018.11.007 |
| [63] |
SU T, WANG W, LI P, ZHANG B, LI P, XIN X, SUN H, YU Y, ZHANG D, ZHAO X, WEN C, ZHOU G, WANG Y, ZHENG H, YU S, ZHANG F. A genomic variation map provides insights into the genetic basis of spring Chinese cabbage (Brassica rapa ssp. pekinensis) selection. Molecular Plant, 2018, 11(11): 1360-1376.
doi: 10.1016/j.molp.2018.08.006 |
| [64] |
SUN J, MA D, TANG L, ZHAO M, ZHANG G, WANG W, SONG J, LI X, LIU Z, ZHANG W, XU Q, ZHOU Y, WU J, YAMAMOTO T, DAI F, LEI Y, LI S, ZHOU G, ZHENG H, XU Z, CHEN W. Population genomic analysis and De Novo assembly reveal the origin of weedy rice as an evolutionary game. Molecular Plant, 2019, 12(5): 632-647.
doi: 10.1016/j.molp.2019.01.019 |
| [65] |
SIMKO I, HAYNES K G, EWING E E, COSTANZO S, CHRIST B J, JONES R W. Mapping genes for resistance to Verticillium alboatrum in tetraploid and diploid potato populations using haplotype association tests and genetic linkage analysis. Molecular Genetics and Genomics, 2004, 271: 522-531.
doi: 10.1007/s00438-004-1010-z |
| [66] |
SCHÖNHALS E M, DING J, RITTER E, PAULO M J, CARA N, TACKE E, HOFFERBERT H R, LÜBECK J, STRAHWALD J, GEBHARDT C. Physical mapping of QTL for tuber yield, starch content and starch yield in tetraploid potato (Solanum tuberosum L.) by means of genome wide genotyping by sequencing and the 8.3 K SolCAP SNP array. BMC Genomics, 2017, 18(1): 642.
doi: 10.1186/s12864-017-3979-9 |
| [67] |
SCHUMACHER C, THÜMECKE S, SCHILLING F, KÖHL K, KOPKA J, SPRENGER H, HINCHA D K, WALTHER D, SEDDIG S, PETERS R, ZUTHER E, HAAS M, HORN R. Genome-wide approach to identify quantitative trait loci for drought tolerance in tetraploid potato (Solanum tuberosum L.). International Journal of Molecular Sciences, 2021, 22(11): 6123.
doi: 10.3390/ijms22116123 |
| [1] | 叶美金, 吴雷, Lohani Md Nahibuzzaman, 尹丽, 胡欣荣, 刘亚西, 蒋云峰, 陈国跃, 蒲至恩, 李阳, 李婷, 邹亚亚, 吴佳怡, 马建. 基于GWAS的中国地方小麦成熟胚大小位点的鉴定及其遗传效应解析[J]. 中国农业科学, 2026, 59(6): 1157-1171. |
| [2] | 杨丽娟, 陈丝雨, 赵薇, 朱玲, 郭磊, 马丽娜, 马瑞敏, 张娟. 全基因组重测序揭示静原鸡羽色的遗传机制[J]. 中国农业科学, 2026, 59(6): 1348-1360. |
| [3] | 王勇胜, 牛丽, 王长杰, 马立花, 廉潇潇, 孟亚雄, 马小乐, 姚立蓉, 张宏, 杨轲, 李葆春, 王化俊, 司二静, 汪军成. 冬小麦千粒重的全基因组关联分析及候选基因预测[J]. 中国农业科学, 2026, 59(3): 499-514. |
| [4] | 何治霖, 孙翠霞, 岳红丽, 谈月霞, 张耀海, 王福生, 刘思涛, 江东. 基于重测序枸橼、柠檬类种质资源的遗传多样性分析和别罗勒烯的全基因组关联分析[J]. 中国农业科学, 2026, 59(2): 386-401. |
| [5] | 李云丽, 刁邓超, 刘雅睿, 孙玉晨, 孟祥宇, 邬陈芳, 汪妤, 吴建辉, 李春莲, 曾庆东, 韩德俊, 郑炜君. 小麦苗期耐热性全基因组关联分析[J]. 中国农业科学, 2025, 58(9): 1663-1683. |
| [6] | 苏明, 李翻过, 洪自强, 周甜, 柳强娟, 班文慧, 吴宏亮, 康建宏. 施氮缓解旱地马铃薯花后高温早衰的抗氧化特性研究[J]. 中国农业科学, 2025, 58(4): 660-675. |
| [7] | 周广飞, 马亮, 马璐, 张舒钰, 章慧敏, 宋旭东, 张振良, 陆虎华, 郝德荣, 冒宇翔, 薛林, 陈国清. 玉米苞叶性状全基因组关联分析[J]. 中国农业科学, 2025, 58(3): 431-442. |
| [8] | 许玉娟, 张捷, 王天译, 陈浩洋, 赵佳佳, 武棒棒, 郝宇琼, 李晓华, 郑兴卫, 左静静, 郑军. 山西小麦低分子量麦谷蛋白Glu-A3和Glu-B3位点多样性鉴定及其对品质的影响[J]. 中国农业科学, 2025, 58(24): 5110-5127. |
| [9] | 马鹤逍, 葛国龙, 张向前, 路战远, 王满秀, 戎美仁, 师晶晶, 张德健, 孙雪萍. 不同作物轮作系统对土壤易氧化有机碳和碳库活度差异性的影响[J]. 中国农业科学, 2025, 58(24): 5201-5215. |
| [10] | 陈彩锦, 马琳, 蒋庆雪, 刘金晖, 苗童, 张志鹏, 孟翔, 马晓冉, 周昕越, 张建, 刘文辉, 王学敏. 244份饲用燕麦种质资源表型性状遗传多样性分析[J]. 中国农业科学, 2025, 58(23): 4825-4836. |
| [11] | 丁宁, 齐恩芳, 贾小霞, 黄伟, 马丽荣, 李建武, 燕汝楠. 马铃薯幼苗应答高温胁迫的miRNA筛选与鉴定[J]. 中国农业科学, 2025, 58(22): 4589-4602. |
| [12] | 魏益民, 周美亮, 唐宇. 荞麦的起源、进化与传播[J]. 中国农业科学, 2025, 58(21): 4305-4316. |
| [13] | 刘晓旭, 钟泽鑫, 邱佳仁, 杨春晓, 张永军, 谢文, 张友军, 潘慧鹏. 豆大蓟马不同地理种群线粒体CO1的遗传多样性[J]. 中国农业科学, 2025, 58(21): 4361-4371. |
| [14] | 武书羽, 衡燕芳, 于太飞, 王世佳, 于思佳, 李园, 胡正, 张辉, 孙现军, 黎亮, 姜奇彦. 玉米自然群体苗期耐盐性鉴定及耐盐相关基因分析[J]. 中国农业科学, 2025, 58(20): 4085-4099. |
| [15] | 唐桂梅, 李卫东, 周宇霞, 孔佑涵, 肖晓玲, 彭颖姝, 张力, 符红艳, 刘洋, 黄国林. 基于表型性状分析蕙兰种质资源遗传多样性[J]. 中国农业科学, 2025, 58(2): 339-354. |
|
||