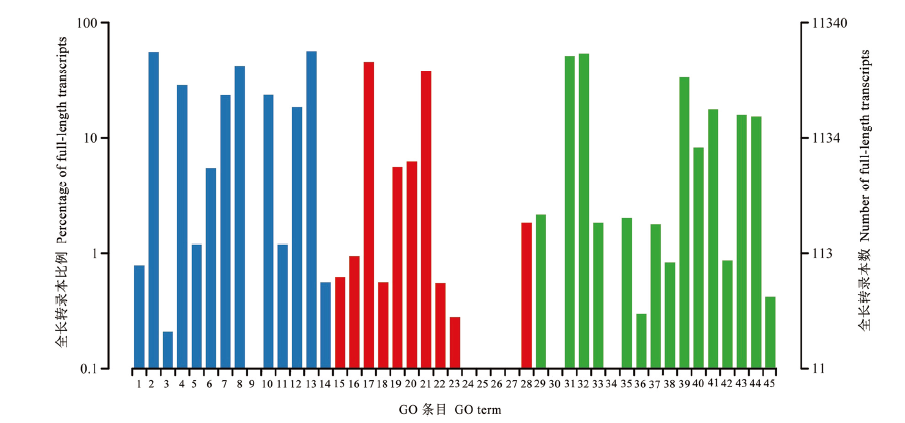
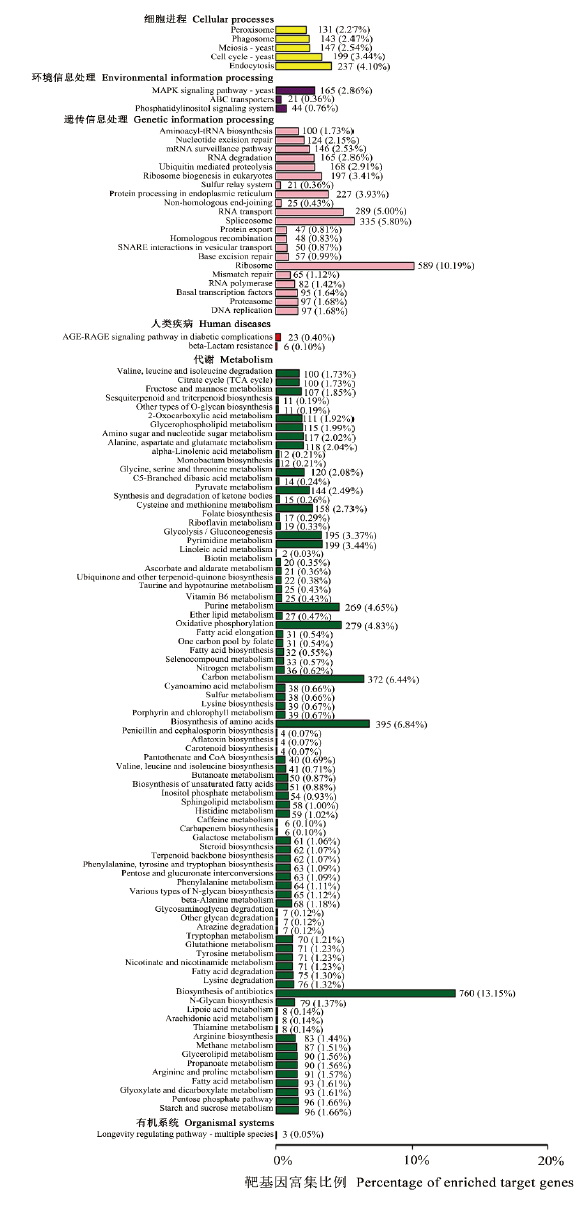
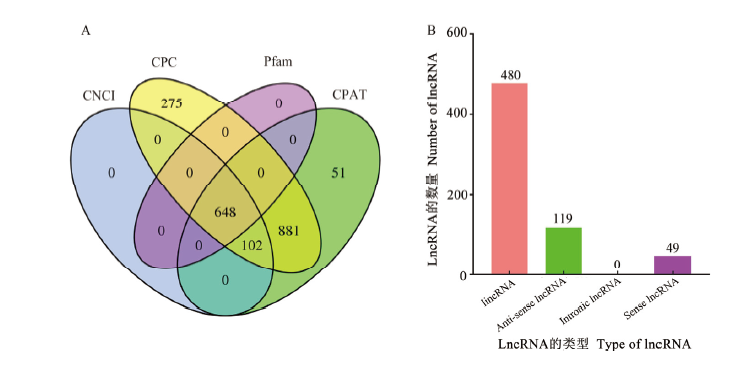
| [1] |
CHEN D F, GUO R, XU X J, XIONG C L, LIANG Q, ZHENG Y Z, LUO Q, ZHANG Z N, HUANG Z J, KUMAR D, XI W J, ZOU X, LIU M. Uncovering the immune responses of Apis mellifera ligustica larval gut to Ascosphaera apis infection utilizing transcriptome sequencing. Gene, 2017,621:40-50.
|
| [2] |
GUO R, CHEN D F, DIAO Q Y, XIONG C L, ZHENG Y Z, HOU C S. Transcriptomic investigation of immune responses of the Apis cerana cerana larval gut infected by Ascosphaera apis. Journal of Invertebrate Pathology, 2019,166:107210.
|
| [3] |
QIN X, EVANS J D, ARONSTEIN K A, MURRAY K D, WEINSTOCK G M. Genome sequences of the honey bee pathogens Paenibacillus larvae and Ascosphaera apis. Insect Molecular Biology, 2006,15(5):715-718.
|
| [4] |
SHANG Y F, XIAO G H, ZHENG P, CEN K, ZHAN S, WANG C S. Divergent and convergent evolution of fungal pathogenicity. Genome Biology and Evolution, 2016,8(5):1374-1387.
|
| [5] |
张曌楠, 熊翠玲, 徐细建, 黄枳腱, 郑燕珍, 骆群, 刘敏, 李汶东, 童新宇, 张琦, 梁勤, 郭睿, 陈大福. 蜜蜂球囊菌的参考转录组de novo组装及SSR分子标记开发. 昆虫学报, 2017,60(1):34-44.
|
|
ZHANG Z N, XIONG C L, XU X J, HUANG Z J, ZHENG Y Z, LUO Q, LIU M, LI W D, TONG X Y, ZHANG Q, LIANG Q, GUO R, CHEN D F. De novo assembly of a reference transcriptome and development of SSR markers for Ascosphaera apis. Acta Entomologica Sinica, 2017,60(1):34-44. (in Chinese)
|
| [6] |
陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 黄枳腱, 张曌楠, 张璐, 李汶东, 童新宇, 席伟军. 胁迫意大利蜜蜂幼虫肠道的球囊菌的转录组分析. 昆虫学报, 2017,60(4):401-411.
|
|
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, HUANG Z J, ZHANG Z N, ZHANG L, LI W D, TONG X Y, XI W J. Transcriptomic analysis of Ascosphaera apis stressing larval gut of Apis mellifera ligustica (Hyemenoptera: Apidae). Acta Entomologica Sinica, 2017,60(4):401-411. (in Chinese)
|
| [7] |
郭睿, 陈大福, 黄枳腱, 梁勤, 熊翠玲, 徐细建, 郑燕珍, 张曌楠, 解彦玲, 童新宇, 侯志贤, 江亮亮, 刀晨. 球囊菌胁迫中华蜜蜂幼虫肠道过程中病原的转录组学研究. 微生物学报, 2017,57(12):1865-1878.
|
|
GUO R, CHEN D F, HUANG Z J, LIANG Q, XIONG C L, XU X J, ZHENG Y Z, ZHANG Z N, XIE Y L, TONG X Y, HOU Z X, JIANG L L, DAO C. Transcriptome analysis of Ascosphaera apis stressing larval gut of Apis cerana cerana. Acta Microbiologica Sinica, 2017,57(12):1865-1878. (in Chinese)
|
| [8] |
郭睿, 李汶东, 陈大福, 熊翠玲, 郑燕珍, 付中民, 徐细建, 黄枳腱, 骆群. 意大利蜜蜂幼虫肠道内球囊菌及其纯培养的高表达基因差异分析. 微生物学通报, 2018,45(2):368-375.
|
|
GUO R, LI W D, CHEN D F, XIONG C L, ZHENG Y Z, FU Z M, XU X J, HUANG Z J, LUO Q. Highly-expressed gene differences between Ascosphaera apis stressing the larval gut of Apis mellifera ligustica and the pure culture of Ascosphaera apis. Microbiology China, 2018,45(2):368-375. (in Chinese)
|
| [9] |
陈大福, 王鸿权, 李汶东, 熊翠玲, 郑燕珍, 付中民, 徐细建, 黄枳腱, 郭睿. 胁迫中华蜜蜂幼虫肠道的球囊菌及其体外培养的高表达基因分析. 福建农林大学学报 (自然科学版), 2017,46(5):562-568.
|
|
CHEN D F, WANG H Q, LI W D, XIONG C L, ZHENG Y Z, FU Z M, XU X J, HUANG Z J, GUO R. Analysis of highly expressed genes of Ascosphaera apis infecting the gut of Apis cerana cerana larvae and its in vitro culture. Journal of Fujian Agriculture and Forestry University (Natural Science Edition), 2017,46(5):562-568. (in Chinese)
|
| [10] |
郭睿, 陈华枝, 童新宇, 熊翠玲, 郑燕珍, 付中民, 解彦玲, 王海朋, 赵红霞, 陈大福. 蜜蜂球囊菌基因结构优化及新基因鉴定. 中国农业大学学报, 2019,24(1):61-68.
|
|
GUO R, CHEN H Z, TONG X Y, XIONG C L, ZHENG Y Z, FU Z M, XIE Y L, WANG H P, ZHAO H X, CHEN D F. Structural optimization of annotated genes and identification of novel genes in Ascosphaera apis. Journal of China Agricultural University, 2019,24(1):61-68. (in Chinese)
|
| [11] |
郭睿, 王海朋, 陈华枝, 熊翠玲, 郑燕珍, 付中民, 赵红霞, 陈大福. 蜜蜂球囊菌的microRNA鉴定及其调控网络分析. 微生物学报, 2018,58(6):1077-1089.
|
|
GUO R, WANG H P, CHEN H Z, XIONG C L, ZHENG Y Z, FU Z M, ZHAO H X, CHEN D F. Identification of Ascosphaera apis microRNAs and investigation of their regulation networks. Acta Microbiologica Sinica, 2018,58(6):1077-1089. (in Chinese)
|
| [12] |
GUO R, CHEN D F, XIONG C L, HOU C S, ZHENG Y Z, FU Z M, DIAO Q Y, ZHANG L, WANG H Q, HOU Z X, LI W D, KUMAR D, LIANG Q. Identification of long non-coding RNAs in the chalkbrood disease pathogen Ascospheara apis. Journal of Invertebrate Pathology, 2018,156:1-5.
|
| [13] |
GUO R, CHEN D F, CHEN H Z, FU Z M, XIONG C L, HOU C S, ZHENG Y Z, GUO Y L, WANG H P, DU Y, DIAO Q Y. Systematic investigation of circular RNAs in Ascosphaera apis, a fungal pathogen of honeybee larvae. Gene, 2018,678:17-22.
|
| [14] |
LU H Y, GIORDANO F, NING Z M. Oxford Nanopore MinION sequencing and genome assembly. Genomics Proteomics and Bioinformatics, 2016,14(5):265-279.
|
| [15] |
WORKMAN R E, TANG A D, TANG P S, JAIN M, TYSON J R, RAZAGHI R, ZUZARTE P C, GILPATRICK T, PAYNE A, QUICK J, et al. Nanopore native RNA sequencing of a human poly (A) transcriptome. Nature Methods, 2019,16(12):1297-1305.
|
| [16] |
LEA W A, PARNELL S C, WALLACE D P, CALVET J P, ZELENCHUK L V, ALVAREZ N S, WARD C J. Human-specific abnormal alternative splicing of wild-type PKD1 induces premature termination of polycystin-1. Journal of the American Society of Nephrology, 2018,29(10):2482-2492.
|
| [17] |
CHEN S Y, DENG F L, JIA X B, LI C, LAI S J. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Scientific Reports, 2017,7:7648.
|
| [18] |
BAYEGA A, OIKONOMOPOULOS S, ZORBAS E, WANG Y C, GREGORIOU M E, TSOUMANI K T, MATHIOPOULOS K D, RAGOUSSIS J. Transcriptome landscape of the developing olive fruit fly embryo delineated by Oxford Nanopore long-read RNA-Seq. bioRxiv, 2018. doi: https://doi.org/10.1101/478172.
|
| [19] |
CHAO Q, GAO Z F, ZHANG D, ZHAO B G, DONG F Q, FU C X, LIU L J, WANG B C. The developmental dynamics of the Populus stem transcriptome. Plant Biotechnology Journal, 2019,17(1):206-219.
|
| [20] |
ZHU C H, LI X F, ZHENG J Y. Transcriptome profiling using Illumina- and SMRT-based RNA-seq of hot pepper for in-depth understanding of genes involved in CMV infection. Gene, 2018,666:123-133.
|
| [21] |
TOMBÁCZ D, BALÁZS Z, CSABAI Z, MOLDOVÁN N, SZŰCS A, SHARON D, SNYDER M, BOLDOGKŐI Z. Characterization of the dynamic transcriptome of a herpesvirus with long-read single molecule real-time sequencing. Scientific Reports, 2017,7:43751.
|
| [22] |
TOMBÁCZ D, BALÁZS Z, CSABAI Z, SNYDER M, BOLDOGKOI Z. Long-read sequencing revealed an extensive transcript complexity in herpesviruses. Frontiers in Genetics, 2018,9:259.
|
| [23] |
陈华枝, 祝智威, 蒋海宾, 王杰, 范元婵, 范小雪, 万洁琦, 卢家轩, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿. 蜜蜂球囊菌菌丝和孢子中微小RNA及其靶mRNA的比较分析. 中国农业科学, 2020,53(17):3606-3619.
|
|
CHEN H Z, ZHU Z W, JIANG H B, WANG J, FAN Y C, FAN X X, WAN J Q, LU J X, XIONG C L, ZHENG Y Z, FU Z M, CHEN D F, GUO R. Comparative analysis of microRNAs and corresponding target mRNAs in Ascospheara apis mycelium and spore. Scientia Agricultura Sinica, 2020,53(17):3606-3619. (in Chinese)
|
| [24] |
陈华枝, 王杰, 祝智威, 蒋海宾, 范元婵, 范小雪, 万洁琦, 卢家轩, 郑燕珍, 付中民, 徐国钧, 陈大福, 郭睿. 蜜蜂球囊菌菌丝和孢子中长链非编码RNA的比较及其潜在功能分析. 中国农业科学, 2021,54(2):435-448.
|
|
CHEN H Z, WANG J, ZHU Z W, JIANG H B, FAN Y C, FAN X X, WAN J Q, LU J X, ZHENG Y Z, FU Z M, XU G J, CHEN D F, GUO R. Comparison and potential functional analysis of long non-coding RNAs between Ascosphaera apis mycelium and spore. Scientia Agricultura Sinica, 2021,54(2):435-448. (in Chinese)
|
| [25] |
CHEN H Z, FAN X X, DU Y, FAN Y C, WANG J, JIANG H B, XIONG C L, ZHENG Y Z, CHEN D F, GUO R. Nanopore-based long-read transcriptome data of Nosema ceranae-infected and un-infected western honeybee workers’ midguts. bioRxiv, 2020. doi: https://doi.org/10.1101/2020.03.21.001958.
|
| [26] |
DU Y, FAN Y C, CHEN H Z, WANG J, XIONG C L, ZHENG Y Z, CHEN D F, GUO R. A full-length transcriptome dataset of normal and Nosema ceranae-challenged midgut tissues of eastern honeybee workers. bioRxiv, 2020. doi: https://doi.org/10.1101/2020.03.18. 997981.
|
| [27] |
JENJAROENPUN P, WONGSURAWAT T, PEREIRA R, PATUMCHAROENPOL P, USSERY D W, NIELSEN J, NOOKAEW I. Complete genomic and transcriptional landscape analysis using third-generation sequencing: A case study of Saccharomyces cerevisiae CEN.PK113-7D. Nucleic Acids Research, 2018,46(7):e38.
|
| [28] |
BOLDOGKOI Z, MOLDOVAN N, BALAZS Z, SNYDER M, TOMBACZ D. Long-read sequencing-A powerful tool in viral transcriptome research. Trends in Microbiology, 2019,27(7):578-592.
|
| [29] |
邓泱泱, 荔建琦, 吴松锋, 朱云平, 陈耀文, 贺福初. nr数据库分析及其本地化. 计算机工程, 2006,32(5):71-73, 76.
|
|
DENG Y Y, LI J Q, WU S F, ZHU Y P, CHEN Y W, HE F C. Integrated nr database in protein annotation system and its localization. Computer Engineering, 2006,32(5):71-73, 76. (in Chinese)
|
| [30] |
The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Research, 2017,45(D1):D158-D169.
|
| [31] |
KOONIN E V, FEDOROVA N D, JACKSON J D, JACOBS A R, KRYLOV D M, MAKAROVA K S, MAZUMDER R, MEKHEDOV S L, NIKOLSKAYA A N, RAO B S, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biology, 2004,5(2):R7.
|
| [32] |
POWELL S, FORSLUND K, SZKLARCZYK D, TRACHANA K, ROTH A, HUERTA-CEPAS J, GABALDÓN T, RATTEI T, CREEVEY C, KUHN M, JENSEN L J, VON MERING C, BORK P. eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Research, 2014,42(Database issue):D231-D239.
|
| [33] |
FINN R D, BATEMAN A, CLEMENTS J, COGGILL P, EBERHARDT R Y, EDDY S R, HEGER A, HETHERINGTON K, HOLM L, MISTRY J, SONNHAMMER E L L, TATE J, PUNTAM. Pfam: The protein families database. Nucleic Acids Research, 2014,42(Database issue):D222-D230.
|
| [34] |
ASHBURNER M, BALL C A, BLAKE J A, BOTSTEIN D, BUTLER H, CHERRY J M, DAVIS A P, DOLINSKI K, DWIGHT S S, EPPIG J T, et al. Gene ontology: Tool for the unification of biology. Nature Genetics, 2000,25(1):25-29.
|
| [35] |
KANEHISA M, GOTO S, KAWASHIMA S, OKUNO Y, HATTORI M. The KEGG resource for deciphering the genome. Nucleic Acids Research, 2004,32(Database issue):D277-D280.
|
| [36] |
熊翠玲, 耿四海, 王心蕊, 刘思亚, 陈大福, 郑燕珍, 付中民, 杜宇, 王海朋, 陈华枝, 周丁丁, 郭睿. 意大利蜜蜂工蜂中肠的长链非编码RNA的预测、分析及鉴定. 应用昆虫学报, 2018,55(6):1034-1044.
|
|
XIONG C L, GENG S H, WANG X R, LIU S Y, CHEN D F, ZHENG Y Z, FU Z M, DU Y, WANG H P, CHEN H Z, ZHOU D D, GUO R. Prediction, analysis and identification of long non-coding RNA in the midguts of Apis mellifera ligustica workers. Chinese Journal of Applied Entomology, 2018,55(6):1034-1044. (in Chinese)
|
| [37] |
KONG L, ZHANG Y, YE Z Q, LIU X Q, ZHAO S Q, WEI L, GAO G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research, 2007,35(Web Server issue):W345-W349.
|
| [38] |
SUN L, LUO H T, BU D C, ZHAO G G, YU K T, ZHANG C H, LIU Y N, CHEN R S, ZHAO Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Research, 2013,41(17):e166.
|
| [39] |
WANG L, PARK H J, DASARI S, WANG S, KOCHER J P, LI W. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Research, 2013,41(6):e74.
|
| [40] |
CHEN D F, DU Y, FAN X X, ZHU Z W, JIANG H B, WANG J, FAN Y C, CHEN H Z, ZHOU D D, XIONG C L, ZHENG Y Z, XU X J, LUO Q, GUO R. Reconstruction and functional annotation of Ascosphaera apis full-length transcriptome via PacBio single-molecule long-read sequencing. bioRxiv, 2019. doi: https://doi.org/10.1101/770040.
|
| [41] |
MAGI A, SEMERARO R, MINGRINO A, GIUSTI B, D’AURIZIO R. Nanopore sequencing data analysis: State of the art, applications and challenges. Briefings in Bioinformatics, 2018,19(6):1256-1272.
|
| [42] |
ARONSTEIN K A, MURRAY K D. Chalkbrood disease in honey bees. Journal of Invertebrate Pathology, 2010,103(Suppl.1):S20-S29.
|
| [43] |
李江红, 郑志阳, 陈大福, 梁勤. 影响蜜蜂球囊菌侵染蜜蜂幼虫的因素及侵染过程观察. 昆虫学报, 2012,55(7):790-797.
|
|
LI J H, ZHENG Z Y, CHEN D F, LIANG Q. Factors influencing Ascosphaera apis infection on honeybee larvae and observation on the infection process. Acta Entomologica Sinica, 2012,55(7):790-797. (in Chinese)
|
| [44] |
TAUBER J P, EINSPANIER R, EVANS J D, MCMAHON D P. Co-incubation of dsRNA reduces proportion of viable spores of Ascosphaera apis, a honey bee fungal pathogen. Journal of Apicultural Research, 2020,59(5):791-799.
|







 ),祝智威1(
),祝智威1(