






中国农业科学 ›› 2020, Vol. 53 ›› Issue (24): 5027-5038.doi: 10.3864/j.issn.0578-1752.2020.24.006
邢启凯1( ),李铃仙1,曹阳2,张玮1,彭军波1,燕继晔1,李兴红1()
),李铃仙1,曹阳2,张玮1,彭军波1,燕继晔1,李兴红1()
收稿日期:2020-02-14
接受日期:2020-03-20
出版日期:2020-12-16
发布日期:2020-12-28
联系方式:
邢启凯,E-mail: qikaixing@163.com。
基金资助:
XING QiKai1(),LI LingXian1,CAO Yang2,ZHANG Wei1,PENG JunBo1,YAN JiYe1,LI XingHong1()
Received:2020-02-14
Accepted:2020-03-20
Published:2020-12-16
Online:2020-12-28
摘要:
【目的】可可毛色二孢(Lasiodiplodia theobromae)是一种世界性分布的重要植物病原真菌,可引起严重的葡萄溃疡病(Botryosphaeria dieback),影响果木品质并造成巨大的经济损失。本研究预测并分析可可毛色二孢基因组范围内的分泌蛋白,并明确其基本特征,为该病菌分泌蛋白致病机理的研究打下基础。【方法】依据已公布的可可毛色二孢全基因组序列,利用信号肽预测软件SignalP v5.0、跨膜结构分析软件TMHMM v2.0、细胞器定位分析软件ProtComp v9.0、GPI锚定预测软件big-PI Fungal Predictor和亚细胞器定位分析软件TargetP v2.0生物信息学软件对该菌中的典型分泌蛋白进行筛选。对分泌蛋白N端信号肽的长度、氨基酸使用频率及其切割位点进行统计分析。依据蛋白序列的同源性,应用BLASTP程序对分泌组蛋白进行功能注释分析,预测其生物学功能。采用蔗糖酶缺陷的酵母分泌系统,对所选分泌蛋白的信号肽进行活性检测。利用qRT-PCR方法检测所选分泌蛋白基因在可可毛色二孢侵染葡萄中的表达情况。【结果】在可可毛色二孢全基因组编码蛋白中共筛选获得552个潜在的具有典型信号肽的分泌蛋白,占全基因组预测蛋白总数的4.3%,其编码蛋白长度集中于101—400 aa。信号肽统计分析表明,其信号肽长度以18—20 aa的序列最为集中,信号肽长度为20 aa的蛋白数量最多。信号肽中使用频率最高的氨基酸为丙氨酸;非极性、疏水的氨基酸使用频率最高,占氨基酸总数的60.2%。其信号肽的-3至-1位置上的氨基酸相对保守,切割位点属于A-X-A类型,可被Sp I型信号肽酶识别并切割。336个分泌蛋白具有功能注释,其功能较多集中于细胞壁降解有关的酶类以及致病相关蛋白,并且这些蛋白在分子量、等电点、脂肪族氨基酸指数等方面均存在差异。通过蔗糖酶缺陷的酵母分泌系统证实,挑选的9个分泌蛋白信号肽均具有分泌活性。qRT-PCR检测结果表明,所选分泌蛋白基因在该病菌侵染初期的表达发生变化。【结论】利用生物信息学分析技术从可可毛色二孢全基因组中共预测获得552个经典分泌蛋白。其信号肽氨基酸长度分布广泛,氨基酸组成中非极性、疏水的氨基酸使用频率最高。功能注释主要集中在细胞壁组分降解相关的酶类、致病侵染相关的坏死诱导相关蛋白以及几丁质结合蛋白等。
邢启凯,李铃仙,曹阳,张玮,彭军波,燕继晔,李兴红. 可可毛色二孢全基因组分泌蛋白的预测及分析[J]. 中国农业科学, 2020, 53(24): 5027-5038.
XING QiKai,LI LingXian,CAO Yang,ZHANG Wei,PENG JunBo,YAN JiYe,LI XingHong. Prediction and Analysis of Candidate Secreted Proteins from the Genome of Lasiodiplodia theobromae[J]. Scientia Agricultura Sinica, 2020, 53(24): 5027-5038.
表1
分泌蛋白信号肽活性测定载体构建引物序列"
| 基因 Gene | 正向引物序列 Forward primer (5′-3′) | 反向引物序列 Reverse primer (5′-3′) |
|---|---|---|
| LT_159 | TTTATGAATTCATGGTCAAGGCTTCCACC | TAATACTCGAGGGCATCGGTGAAGGTGCAG |
| LT_188 | TTTATGAATTCATGCGTGTTTCGACTCTTC | TAATACTCGAGAAAGAAGGTGAAGGTAGAAG |
| LT_233 | TTTATGAATTCATGGTCAAGGTTTCCACC | TAATACTCGAGGGTGAAAGTGCAGCTGG |
| LT_359 | TTTATGAATTCATGCCTTCCCTCAAGTC | TAATACTCGAGGTTTTCGGCGGCCTGGG |
| LT_595 | TACAGGAATTCATGCGTTCCTCTGCTC | GACTGCTCGAGCACGATGTCGAGATCAG |
| LT_62 | CCGGAATTCATGGGCTGGTTTTGGTTC | CCGCTCGAGCACGACGGTGATCGTCG |
| LT_936 | TACTAGAATTCATGAAGGCTTCCGGTC | CACTTCTCGAGACCGTTGACAGCCTGAC |
| LT_1541 | TTTATGAATTCATGGTGTCCTTCCGCTCTC | TAATACTCGAGGAGAGACTGCCTGGCAATC |
| LT_1698 | TTTATGAATTCATGAAGTTCTCTACCACC | TAATACTCGAGGTCCTCGGTGACCTCGCC |
表2
实时荧光定量PCR所用引物序列"
| 基因 Gene | 正向引物序列 Forward primer (5′-3′) | 反向引物序列 Reverse primer (5′-3′) |
|---|---|---|
| LT_159 | CCAGCAGGACTACAAGAA | CCAGAGGTAGACCAGTTC |
| LT_188 | CTACCTTGCCGACCTTAA | GATGATGTTGCCGTTGAA |
| LT_233 | GAGCAGGACTACGAGAAC | CGCAGAGGATGTAGATGT |
| LT_359 | CAAGTCTTCCTCCATCCA | GATCTGAGCCGAGTTGTA |
| LT_595 | AGATGGTCTGGAAGAACTC | CGTACTCGTCAAGGATGT |
| LT_62 | GGAATCAACGACGACTCT | CGCACTGTGTTGGTTATG |
| LT_936 | CTACAACGAAATCAGCGAAT | ATGGTGGTGGTCTTCTTC |
| LT_1541 | CAACGGCTACTACTACTCTT | TTGATGTTCCTGGCACTG |
| LT_1698 | AATGGTGCTCAGTTCTACA | AGATGTTGATGAGGAGACC |
| LT_Actin | TCTTCGCTCGAGAAGTCGTA | ACAATGGAAGGTCCGCTCTC |
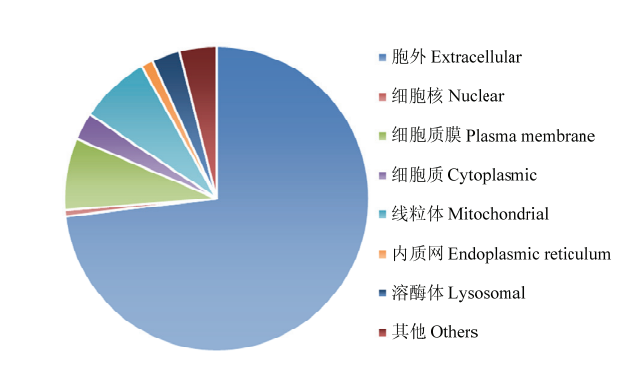
图1
可可毛色二孢中937个具有信号肽蛋白的亚细胞定位"
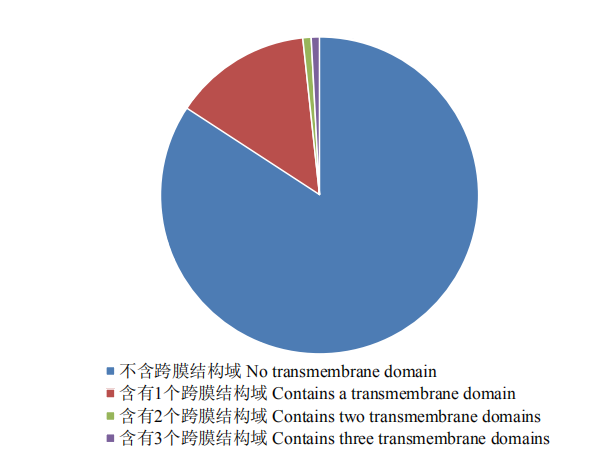
图2
可可毛色二孢中685个外泌蛋白的跨膜结构域分析"
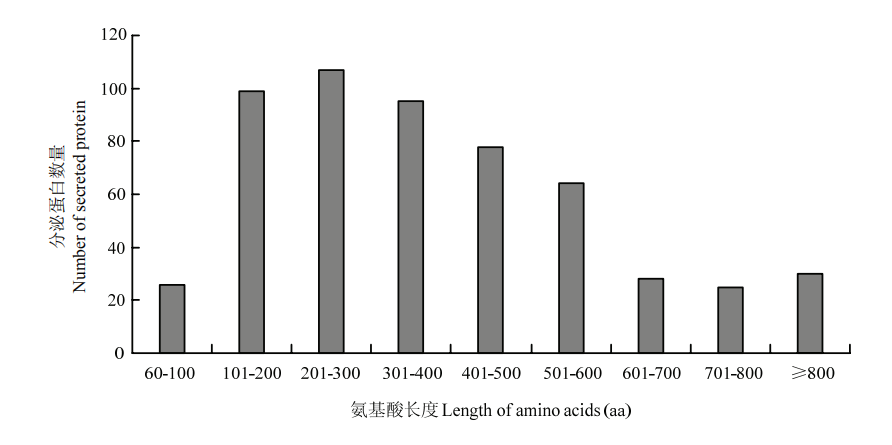
图3
可可毛色二孢典型分泌蛋白的序列长度"

图4
可可毛色二孢分泌蛋白的信号肽长度分布"
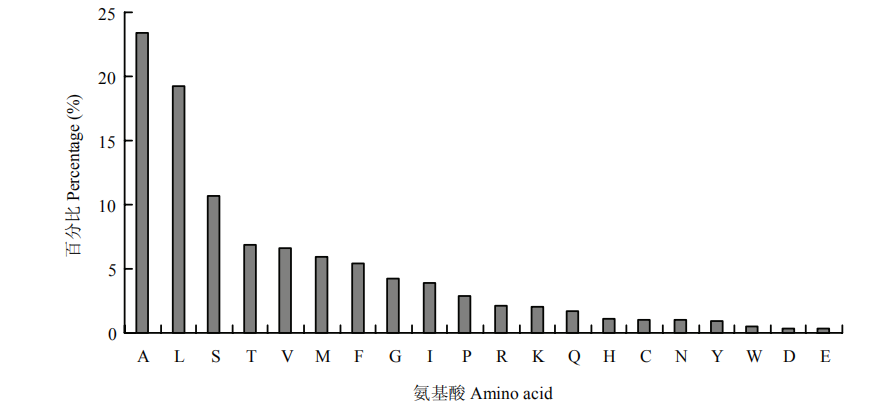
图5
可可毛色二孢分泌蛋白氨基酸使用频率"
表3
可可毛色二孢分泌蛋白信号肽切割位点的氨基酸组成分布"
| 氨基酸类型 Type of amino acids | 信号肽切割位点-3到3位的氨基酸组成 Frequency of amino acids from -3 to +3 at signal peptide cleavage site of secreted proteins | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| -3 | -2 | -1 | 1 | 2 | 3 | |||||||
| 数量 Amount | 百分比 Percentage (%) | 数量 Amount | 百分比 Percentage (%) | 数量 Amount | 百分比 Percentage (%) | 数量 Amount | 百分比 Percentage (%) | 数量 Amount | 百分比 Percentage (%) | 数量 Amount | 百分比 Percentage (%) | |
| A | 249 | 47.0 | 60 | 11.3 | 387 | 73.0 | 145 | 27.3 | 26 | 4.9 | 35 | 6.6 |
| C | 13 | 2.5 | 6 | 1.1 | 2 | 0.4 | 18 | 3.4 | 6 | 1.1 | 21 | 4.0 |
| D | 2 | 0.4 | 9 | 1.7 | 3 | 0.6 | 38 | 7.2 | 51 | 9.6 | 19 | 3.6 |
| E | 8 | 1.5 | 12 | 2.3 | 2 | 0.4 | 24 | 4.5 | 35 | 6.6 | 16 | 3.0 |
| F | 1 | 0.2 | 26 | 4.9 | 5 | 0.9 | 12 | 2.3 | 6 | 1.1 | 20 | 3.8 |
| G | 16 | 3.0 | 10 | 1.9 | 19 | 3.6 | 20 | 3.8 | 23 | 4.3 | 23 | 4.3 |
| H | 1 | 0.2 | 29 | 5.5 | 0 | 0 | 20 | 3.8 | 3 | 0.6 | 12 | 2.3 |
| I | 10 | 1.9 | 10 | 1.9 | 0 | 0 | 12 | 2.3 | 7 | 1.3 | 38 | 7.2 |
| K | 0 | 0 | 3 | 0.6 | 8 | 1.5 | 19 | 3.6 | 3 | 0.6 | 8 | 1.5 |
| L | 22 | 4.2 | 87 | 16.4 | 9 | 1.7 | 21 | 4.0 | 15 | 1.3 | 49 | 9.2 |
| M | 0 | 0 | 10 | 1.9 | 1 | 0.2 | 4 | 0.8 | 2 | 0.4 | 4 | 0.8 |
| N | 1 | 0.2 | 22 | 4.2 | 5 | 0.9 | 14 | 2.6 | 21 | 4.0 | 22 | 4.2 |
| P | 8 | 1.5 | 7 | 1.3 | 22 | 4.2 | 5 | 0.9 | 148 | 27.9 | 44 | 8.3 |
| Q | 5 | 0.9 | 48 | 9.1 | 6 | 1.1 | 68 | 12.8 | 24 | 4.5 | 25 | 4.7 |
| R | 9 | 1.7 | 27 | 5.1 | 3 | 0.6 | 10 | 1.9 | 6 | 1.1 | 14 | 2.6 |
| S | 56 | 10.6 | 68 | 12.8 | 36 | 6.8 | 30 | 5.7 | 44 | 8.3 | 40 | 7.5 |
| T | 40 | 7.5 | 44 | 8.3 | 9 | 1.7 | 26 | 4.9 | 61 | 11.5 | 74 | 14.0 |
| V | 89 | 16.8 | 35 | 6.6 | 9 | 1.7 | 31 | 5.8 | 33 | 6.2 | 41 | 7.7 |
| W | 0 | 0 | 1 | 0.2 | 1 | 0.2 | 4 | 0.8 | 6 | 1.1 | 5 | 0.9 |
| Y | 0 | 0 | 16 | 3.0 | 3 | 0.6 | 9 | 1.7 | 10 | 1.9 | 20 | 3.8 |
表4
可可毛色二孢部分分泌蛋白生化特性与功能注释"
| 基因编号 Gene ID | 蛋白长度 Length (aa) | 分子量 Molecular weight (kD) | 等电点 pI | 脂肪族氨基酸指数 Aliphatic index | 功能注释 Function |
|---|---|---|---|---|---|
| evm.model.scaffold_1.1884 | 265 | 28.57 | 4.57 | 70.38 | 糖基水解酶Glycosyl hydrolases family |
| evm.model.scaffold_7.188 | 334 | 36.85 | 4.62 | 71.89 | 纤维素酶Cellulase |
| evm.model.scaffold_6.309 | 322 | 36.16 | 5.96 | 80.90 | 水解酶家族Alpha/beta hydrolase family |
| evm.model.scaffold_3.829 | 549 | 59.13 | 5.16 | 82.00 | 羧酸酯酶Carboxylesterase family |
| evm.model.scaffold_5.474 | 324 | 35.76 | 4.96 | 83.36 | 过氧化物酶Peroxidase |
| evm.model.scaffold_5.945 | 348 | 39.24 | 5.23 | 69.20 | 酪氨酸酶Tyrosinase |
| evm.model.scaffold_13.28 | 251 | 26.69 | 5.34 | 82.03 | 角质酶Cutinase |
| evm.model.scaffold_6.540 | 395 | 42.03 | 5.39 | 74.63 | 天冬氨酸蛋白酶Aspartyl protease |
| evm.model.scaffold_8.302 | 265 | 27.76 | 5.55 | 65.92 | 脂肪酶GDSL-like Lipase |
| evm.model.scaffold_4.1035 | 581 | 62.25 | 4.80 | 87.04 | 氧化还原酶GMC oxidoreductase |
| evm.model.scaffold_3.522 | 252 | 26.19 | 4.13 | 74.64 | 果胶酸裂解酶Pectate lyase |
| evm.model.scaffold_2.202 | 377 | 40.24 | 5.22 | 73.37 | 肽酶Peptidase family |
| evm.model.scaffold_2.1494 | 201 | 20.37 | 4.41 | 60.45 | WSC结构域蛋白WSC domain protein |
| evm.model.scaffold_11.25 | 430 | 45.29 | 4.01 | 63.14 | PAN结构域蛋白PAN domain protein |
| evm.model.scaffold_1.947 | 254 | 28.05 | 8.38 | 57.01 | 坏死诱导蛋白Necrosis inducing protein |
| evm.model.scaffold_4.1274 | 186 | 19.83 | 4.43 | 61.51 | LysM结构域蛋白LysM domain protein |
| evm.model.scaffold_10.213 | 122 | 12.15 | 4.33 | 111.15 | FAD结构域蛋白FAD domain protein |
| evm.model.scaffold_1.937 | 247 | 26.14 | 5.05 | 84.45 | Cupin结构域蛋白Cupin domain protein |
| evm.model.scaffold_14.112 | 246 | 23.24 | 3.90 | 71.14 | CFEM结构域蛋白CFEM domain protein |
| evm.model.scaffold_11.308 | 412 | 40.74 | 5.14 | 53.98 | 几丁质结合蛋白Chitin binding protein |

图6
分泌蛋白信号肽功能验证"
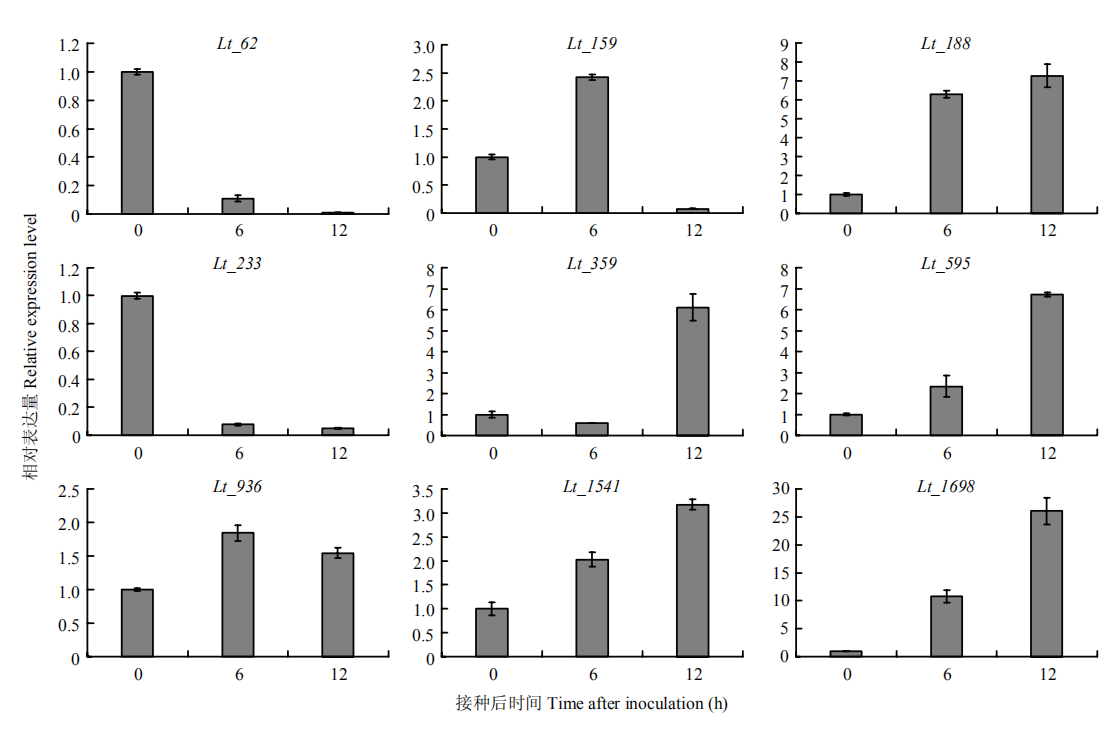
图7
9个候选分泌蛋白基因在可可毛色二孢侵染过程中的相对表达量"
| [1] |
BERTSH C, LARIGNON P, FARINE S, CLEMENT C, FONTAINE F. The spread of grapevine trunk disease. Science, 2009,324(5928):721.
doi: 10.1126/science.324_721b pmid: 19423798 |
| [2] | YAN J Y, XIE Y, YAO S W, WANG Z Y, LI X H. Characterization of Botryosphaeria dothidea, the causal agent of grapevine canker in China. Australasian Plant Pathology, 2012,41(4):351-357. |
| [3] | YAN J Y, XIE Y, ZHANG W, WANG Y, LIU J K, HYDE K D, SEEM R C, ZHANG G Z, WANG Z Y, YAO S W, BAI X J, DISSANAYAKE A J, PENG Y L, LI X H. Species of Botryosphaeriaceae involved in grapevine dieback in China. Fungal Diversity, 2013,61(1):221-236. |
| [4] | DISSANAYAKE A J, ZHANG W, LIU M, CHUKEATIROTE E, YAN J Y, LI X H, HYDE K D. Lasiodiplodia pseudotheobromae causes pedicel and peduncle discolouration of grapes in China. Australasian Plant Disease Notes, 2015,10:21. |
| [5] | DISSANAYAKE A J, ZHANG W, LI X H, ZHOU Y, CHETHANA T, CHUKEATIROTE E, HYDE K D, YAN J Y, ZHANG G Z, ZHAO W S. First report of Neofusicoccum mangiferae associated with grapevine dieback in China. Phytopathologia Mediterranea, 2015,54(2):414-419. |
| [6] |
NIMCHUK Z, EULGEM T, HOLT III B F, DANGL J L. Recognition and response in the plant immune system. Annual Review of Genetics, 2003,37:579-609.
pmid: 14616074 |
| [7] | RODRIGUEZ-MORENO L, EBERT M K, BOLTON M D, THOMMA B P H J. Tools of the crook-infection strategies of fungal plant pathogens. The Plant Journal, 2018,93(4):664-674. |
| [8] | GREENBAUM D, LUSCOMBE N M, JANSEN R, QIAN J, GERSTEIN M. Interrelating different types of genomic data, from proteome to secretome: ′Oming in on function. Genome Research, 2001,11(9):1463-1468. |
| [9] |
DE SAIN M, REP M. The role of pathogen-secreted proteins in fungal vascular wilt diseases. International Journal of Molecular Sciences, 2015,16(10):23970-23993.
pmid: 26512660 |
| [10] |
CHOI J, PARK J, KIM D, JUNG K, KANG S, LEE Y H. Fungal secretome database: Integrated platform for annotation of fungal secretomes. BMC Genomics, 2010,11:105.
doi: 10.1186/1471-2164-11-105 |
| [11] |
VAN DER BURGH A M, JOOSTEN M H. Plant immunity: Thinking outside and inside the box. Trends in Plant Science, 2019,24(7):587-601.
pmid: 31171472 |
| [12] |
JONES J D, DANGL J L. The plant immune system. Nature, 2006,444:323-329.
doi: 10.1038/nature05286 pmid: 17108957 |
| [13] |
OLIVEIRA-GARCIA E, VALENT B. How eukaryotic filamentous pathogens evade plant recognition. Current Opinion in Microbiology, 2015,26:92-101.
pmid: 26162502 |
| [14] |
ASAI S, SHIRASU K. Plant cells under siege: Plant immune system versus pathogen effectors. Current Opinion in Plant Biology, 2015,28:1-8.
pmid: 26343014 |
| [15] |
FRANCESCHETTI M, MAQBOOL A, JIMENEZ-DALMARONI M J, PENNINGTON H G, KAMOUN S, BANFIELD M J. Effectors of filamentous plant pathogens: Commonalities amid diversity. Microbiology and Molecular Biology Reviews, 2017,81(2):e00066-16.
pmid: 28356329 |
| [16] |
田李, 陈捷胤, 陈相永, 汪佳妮, 戴小枫. 大丽轮枝菌 (Verticillium dahliae VdLs. 17) 分泌组预测及分析. 中国农业科学, 2011,44(15):3142-3153.
doi: 10.3864/j.issn.0578-1752.2011.15.009 |
|
TIAN L, CHEN J Y, CHEN X Y, WANG J N, DAI X F. Prediction and analysis of Verticillium dahliae VdLs. 17 secretome. Scientia Agricultura Sinica, 2011,44(15):3142-3153. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2011.15.009 |
|
| [17] | 周晓罡, 侯思名, 陈铎文, 陶南, 丁玉梅, 孙茂林, 张绍松. 马铃薯晚疫病菌全基因组分泌蛋白的初步分析. 遗传, 2011,33(7):785-793. |
| ZHOU X G, HOU S M, CHEN D W, TAO N, DING Y M, SUN M L, ZHANG S S. Genome-wide analysis of the secreted proteins of Phytophthora infestans. Hereditas, 2011,33(7):785-793. (in Chinese) | |
| [18] | 陈琦光, 王陈骄子, 杨媚, 周而勋. 希金斯刺盘孢全基因组候选效应分子的预测. 热带作物学报, 2015,36(6):1105-1111. |
| CHEN Q G, WANG C J Z, YANG M, ZHOU E X. Prediction of candidate effectors from the genome of Colletotrichum higginsianum. Chinese Journal of Tropical Crops, 2015,36(6):1105-1111. (in Chinese) | |
| [19] | 韩长志. 全基因组预测禾谷炭疽菌的分泌蛋白. 生物技术, 2014,24(2):36-41. |
| HAN C Z. Prediction for secreted proteins from Colletotrichum graminicola genome. Biotechnology, 2014,24(2):36-41. (in Chinese) | |
| [20] |
DE CARVALHO M C, NASCIMENTO L C, DARBEN L M, POLIZEL- PODANOSQUI A M, LOPES-CAITAR V S, QI M, ROCHA C S, CARAZZOLLE M F, KUWAHARA M K, PEREIRA G A, ABDELNOOR R V, WHITHAM S A, MARCELINO-GUIMARAES F C. Prediction of the in planta Phakopsora pachyrhizi secretome and potential effector families. Molecular Plant Pathology, 2017,18(3):363-377.
doi: 10.1111/mpp.12405 pmid: 27010366 |
| [21] | ZENG R, GAO S G, XU L X, LIU X, DAI F M. Prediction of pathogenesis-related secreted proteins from Stemphylium lycopersici. BMC Microbiology, 2018,18(1):191. |
| [22] |
YAN J Y, ZHAO W S, CHEN Z, XING Q K, ZHANG W, CHETHANA K W T, XUE M F, XU J P, PHILLIPS A J L, WANG Y, et al. Comparative genome and transcriptome analyses reveal adaptations to opportunistic infections in woody plant degrading pathogens of Botryosphaeriaceae. DNA Research, 2018,25(1):87-102.
doi: 10.1093/dnares/dsx040 pmid: 29036669 |
| [23] | ARMENTEROS J J A, TSIRIGOS K D, SONDERBY C K, PETERSEN T N, WINTHER O, BRUNAK S, VON HEIJNE G, NIELSEN H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology, 2019,37(4):420-423. |
| [24] | EMANUELSSON O, NIELSEN H, BRUNAK S, VON HEIJNE G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. Journal of Molecular Biology, 2000,300(4):1005-1016. |
| [25] |
KROGH A, LARSSON B E, VON HEIJNE G, SONNHAMMER E L L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. Journal of Molecular Biology, 2001,305(3):567-580.
doi: 10.1006/jmbi.2000.4315 pmid: 11152613 |
| [26] |
EISENHABER B, BORK P, EISENHABER F. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: Analysis of protein sequence data from complete genomes. Protein Engineering, 2001,14(1):17-25.
pmid: 11287675 |
| [27] |
ARMENTEROS J J A, SALVATORE M, EMANUELSSON O, WINTHER O, VON HEIJNE G, ELOFSSON A, NIELSEN H. Detecting sequence signals in targeting peptides using deep learning. Life Science Alliance, 2019,2(5):e201900429.
pmid: 31515291 |
| [28] |
JUNCKER A S, WILLENBROCK H, VON HEIJNE G, NIELSEN H, BRUNAK S, KROGH A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Science, 2003,12(8):1652-1662.
pmid: 12876315 |
| [29] | GASTEIGER E, HOOGLAND C, GATTIKER A, DUVAUD S, WILKINS M R, APPEL R D, BAIROCH A. Protein identification and analysis tools on the ExPASy server//The Proteomics Protocols Handbook. Humana Press, 2005: 571-607. |
| [30] | JACOBS K A, COLLINS-RACIE L A, COLBERT M, DUCKETT M, GOLDEN-FLEET M, KELLEHER K, KRIZ R, LAVALLIE E R, MERBERG D, SPAULDING V, STOVER J, WILLIAMSON M J, MCCOY J M. A genetic selection for isolating cDNAs encoding secreted proteins. Gene, 1997,198(1/2):289-296. |
| [31] |
FANG A F, HAN Y Q, ZHANG N, ZHANG M, LIU L J, LI S, LU F, SUN W X. Identification and characterization of plant cell death- inducing secreted proteins from Ustilaginoidea virens. Molecular Plant-Microbe Interactions, 2016,29(5):405-416.
doi: 10.1094/MPMI-09-15-0200-R pmid: 26927000 |
| [32] | SONAH H, DESHMUKH R K, BELANGER R R. Computational prediction of effector proteins in fungi: Opportunities and challenges. Frontiers in Plant Science, 2016,7:126. |
| [33] |
WAN W L, FROHLICH K, PRUITT R N, NURNBERGER T, ZHANG L. Plant cell surface immune receptor complex signaling. Current Opinion in Plant Biology, 2019,50:18-28.
doi: 10.1016/j.pbi.2019.02.001 pmid: 30878771 |
| [34] | TORUNO T Y, STERGIOPOULOS I, COAKER G. Plant-pathogen effectors: Cellular probes interfering with plant defenses in spatial and temporal manners. Annual Review of Phytopathology, 2016,54:419-441. |
| [35] |
LIU T, SONG T, ZHANG X, YUAN H, SU L, LI W, XU J, LIU S, CHEN L, CHEN T, ZHANG M, GU L, ZHANG B, DOU D. Unconventionally secreted effectors of two filamentous pathogens target plant salicylate biosynthesis. Nature Communications, 2014,5:4686.
pmid: 25156390 |
| [36] | 李云锋, 聂燕芳, 王振中. 植物病原真菌分泌蛋白质组学研究进展. 微生物学通报, 2015,42(6):1101-1107. |
| LI Y F, NIE Y F, WANG Z Z. Research progress on secretomics of phytopathogenic fungi. Microbiology China, 2015,42(6):1101-1107. (in Chinese) | |
| [37] |
BROUWER H, COUTINHO P M, HENRISSAT B, DE VRIES R P. Carbohydrate-related enzymes of important Phytophthora plant pathogens. Fungal Genetics and Biology, 2014,72:192-200.
doi: 10.1016/j.fgb.2014.08.011 pmid: 25192612 |
| [38] |
SANCHEZ-VALLET A, MESTERS J R, THOMMA B P. The battle for chitin recognition in plant-microbe interactions. FEMS Microbiology Reviews, 2015,39(2):171-183.
doi: 10.1093/femsre/fuu003 pmid: 25725011 |
| [39] |
AKCAPINAR G B, KAPPEL L, SEZERMAN O U, SEIDL-SEIBOTH V. Molecular diversity of LysM carbohydrate-binding motifs in fungi. Current Genetics, 2015,61(2):103-113.
pmid: 25589417 |
| [1] | 江峰, 吴春燕, 王亦好, 杨泽众, 龚成, 罗晨. 烟粉虱MED脂肪酸延长酶基因家族鉴定和表达分析[J]. 中国农业科学, 2026, 59(4): 793-806. |
| [2] | 韦萍, 潘炬忠, 朱德平, 邵胜雪, 陈珊珊, 韦雅倩, 高维维. OsDREB1J调控水稻籽粒大小的功能研究[J]. 中国农业科学, 2025, 58(8): 1463-1478. |
| [3] | 刘路平, 胡雪洁, 祁金, 陈强, 刘智, 赵田湉, 史晓蕾, 刘兵强, 孟庆民, 张孟臣, 韩天富, 杨春燕. 大豆生育期基因E1和E2的启动子克隆及其表达模式分析[J]. 中国农业科学, 2025, 58(5): 840-850. |
| [4] | 张天雨, 李白, 藏金萍, 曹宏哲, 董金皋, 邢继红, 张康. 灰葡萄孢HMG家族基因的全基因组鉴定与表达规律分析[J]. 中国农业科学, 2025, 58(4): 704-718. |
| [5] | 仪泽会, 王颖, 宋慧霞, 赵婧, 毛丽萍. 芦笋过氧化物还原酶(Peroxiredoxins)基因家族全基因组鉴定及表达分析[J]. 中国农业科学, 2025, 58(18): 3728-3743. |
| [6] | 王伟, 吴传磊, 胡晓渝, 李佳佳, 白鹏宇, 王郭伋, 苗龙, 王晓波. 大豆LOX基因家族的全基因组鉴定及GmLOX15A1基因等位变异对百粒重的影响[J]. 中国农业科学, 2025, 58(1): 10-29. |
| [7] | 冯雯蜜, 周芳雪, 于哲, 牟可欣, 井妍, 李海燕. 大豆抗花叶病毒病基因GmRHF1的克隆及功能分析[J]. 中国农业科学, 2024, 57(23): 4632-4643. |
| [8] | 吴传磊, 胡晓渝, 王伟, 苗龙, 白鹏宇, 王郭伋, 李娜, 舒阔, 邱丽娟, 王晓波. 大豆油分相关功能基因分子标记开发与鉴定及优异等位变异聚合分析[J]. 中国农业科学, 2024, 57(22): 4402-4415. |
| [9] | 孙思思, 马伍, 司惠茹, 王贤忠, 柳强, 罗炎琳, 陈孝玉龙, 唐斌. 豌豆修尾蚜水通道蛋白特征及响应高湿度胁迫表达变化[J]. 中国农业科学, 2024, 57(20): 4057-4070. |
| [10] | 潘凤英, 曲俊杰, 刘露露, 孙大运, 郭泽西, 韦晓丽, 韦淑梅, 尹玲. 葡萄霜霉菌糖基水解酶基因的表达模式与功能分析[J]. 中国农业科学, 2023, 56(5): 879-891. |
| [11] | 张开京, 何帅帅, 贾利, 胡玉超, 杨德坤, 陆晓民, 张其安, 严从生. 黄瓜DIR家族基因的全基因组鉴定及其表达分析[J]. 中国农业科学, 2023, 56(4): 711-728. |
| [12] | 党媛玥, 马建江, 杨书贤, 宋吉坤, 贾冰, 冯盼, 陈全家, 于霁雯. 棉花β-tubulin基因家族的鉴定及其在纤维发育中的表达[J]. 中国农业科学, 2023, 56(23): 4585-4601. |
| [13] | 董琰钰, 徐碧玉, 董泽宇, 汪露瑶, 陈锦文, 方磊. 棉花EXO70基因家族全基因组的鉴定及种间比较[J]. 中国农业科学, 2023, 56(23): 4621-4634. |
| [14] | 韩晓文, 韩硕, 胡义锋, 王梦如, 陈中义, 朱永兴, 尹军良. 喜旱莲子草AP2/ERF基因家族的鉴定及其在除草剂胁迫下的表达模式[J]. 中国农业科学, 2023, 56(20): 4021-4034. |
| [15] | 丁国华, 肖光辉, 竺丽萍. 棉花NLP(NIN-Like Protein)基因家族的全基因组鉴定及表达分析[J]. 中国农业科学, 2023, 56(19): 3723-3746. |
|
||