






中国农业科学 ›› 2020, Vol. 53 ›› Issue (9): 1704-1716.doi: 10.3864/j.issn.0578-1752.2020.09.002
所属专题: 专题——限制性两阶段多位点全基因组关联分析法的应用
• 专题:限制性两阶段多位点全基因组关联分析法的应用 • 上一篇 下一篇
贺建波,刘方东,王吴彬,邢光南,管荣展,盖钧镒( )
)
收稿日期:2019-08-26
接受日期:2019-11-30
出版日期:2020-05-01
发布日期:2020-05-13
联系方式:
贺建波,E-mail:hjbxyz@gmail.com。
基金资助:
JianBo HE,FangDong LIU,WuBin WANG,GuangNan XING,RongZhan GUAN,JunYi GAI()
Received:2019-08-26
Accepted:2019-11-30
Published:2020-05-01
Online:2020-05-13
摘要:
全基因组关联分析(genome-wide association studies,GWAS)通过建立全基因组高密度分子标记以检测基因型与表型间的关联性,已成为动植物数量性状遗传解析的主要方法。然而,以往GWAS方法只注重于个别主要QTL的检测,而且使用仅有2个等位变异的SNP标记不能检测自然群体中广泛存在的复等位变异,一定程度限制了GWAS的应用。限制性两阶段多位点全基因组关联分析方法(RTM-GWAS)首先根据全基因组高密度SNP标记间的连锁不平衡程度,将多个相邻且紧密连锁的SNP标记组成为具有复等位变异(单倍型)的连锁不平衡区段(SNPLDB)标记。其次,RTM-GWAS使用由SNPLDB标记计算的遗传相似系数矩阵作为群体结构偏差的通用估计,并提取该矩阵的特征向量作为模型协变量以降低由群体结构偏差导致的假阳性。最后,利用具有复等位变异的SNPLDB标记与建立的多位点复等位变异模型,RTM-GWAS将性状遗传率作为QTL表型变异解释率的上限,通过两阶段分析策略高效地进行全基因组QTL及其复等位变异的检测,并最终构建多QTL遗传模型。该法还可以基于性状小区观测值,建立QTL与环境互作多位点模型,不仅能检测与环境有交互作用的主效应QTL,还能检测仅与环境有交互作用的无主效应QTL。RTM-GWAS不仅解决了以往GWAS不能估计复等位变异的问题,而且通过使用多位点模型拟合多个QTL提高了检测功效并能有效地控制假阳性的膨胀,为全面解析自然群体QTL及其复等变异提供了通道。该法能估计出等位基因的效应及其在群体内的相对频率,由其结果建立的QTL-allele矩阵代表了目标性状在群体中的全部遗传组成,不仅可用于候选基因发掘,还为群体内QTL及其复等位变异(基因及其复等位基因)的动态研究(群体遗传分化以及特有与新生等位变异)提供了新的工具。依据QTL-allele矩阵,还能进一步利用计算机模拟产生杂交组合后代基因型,并预测杂交组合后代纯合群体的表现,从而进行优化组合设计与分子设计育种。此外,RTM-GWAS还适用于双亲杂交后代重组自交系群体以及多亲杂交后代巢式关联作图群体,因避免了群体结构偏离的干扰,检测功效更高。本文归纳了RTM-GWAS的原理和方法,并综述了其在遗传育种研究中的应用。
贺建波,刘方东,王吴彬,邢光南,管荣展,盖钧镒. 限制性两阶段多位点全基因组关联分析法在遗传育种中的应用[J]. 中国农业科学, 2020, 53(9): 1704-1716.
JianBo HE,FangDong LIU,WuBin WANG,GuangNan XING,RongZhan GUAN,JunYi GAI. Restricted Two-Stage Multi-Locus Genome-Wide Association Analysis and Its Applications to Genetic and Breeding Studies[J]. Scientia Agricultura Sinica, 2020, 53(9): 1704-1716.
表1
中国大豆种质资源群体百粒重显著关联的SNPLDB标记位点"
| SNPLDB | 染色体 Chromosome | 物理位置Position (bp) a | 等位基因数目 No. alleles | -lgP | R2 (%) |
|---|---|---|---|---|---|
| LDB_18_59996683 | 18 | 59996683 | 2 | 129.8 | 9.84 |
| LDB_8_5286591 | 8 | 5286591 | 2 | 99.3 | 6.76 |
| LDB_16_35761014 | 16 | 35761014—35771300 | 4 | 86.0 | 5.80 |
| LDB_6_3703919 | 6 | 3703919 | 2 | 84.1 | 5.43 |
| LDB_4_3019467 | 4 | 3019467—3046646 | 3 | 81.7 | 5.35 |
| LDB_17_15063207 | 17 | 15063207—15063454 | 4 | 61.0 | 3.80 |
| LDB_11_28584788 | 11 | 28584788—28784681 | 8 | 53.6 | 3.53 |
| LDB_14_47245011 | 14 | 47245011 | 2 | 37.9 | 2.08 |
| LDB_9_6122236 | 9 | 6122236 | 2 | 37.5 | 2.05 |
| LDB_13_42639761 | 13 | 42639761 | 2 | 36.5 | 1.99 |
| ... | ... | ... | ... | ... | ... |
| LDB_2_11741211 | 2 | 11741211—11741518 | 3 | 9.1 | 0.47 |
| LDB_10_34650810 | 10 | 34650810—34706889 | 5 | 7.6 | 0.46 |
| LDB_5_38249682 | 5 | 38249682—38278658 | 5 | 7.3 | 0.45 |
| LDB_7_35863030 | 7 | 35863030—35901005 | 6 | 6.9 | 0.45 |
| LDB_9_1954783 | 9 | 1954783 | 2 | 9.4 | 0.44 |
| ... | ... | ... | ... | ... | ... |
| LDB_8_44667459 | 8 | 44667459 | 2 | 2.6 | 0.10 |
| LDB_13_35141544 | 13 | 35141544 | 2 | 2.6 | 0.10 |
| LDB_18_61536415 | 18 | 61536415 | 2 | 2.7 | 0.10 |
| ... | ... | ... | ... | ... | ... |
| LDB_8_16362965 | 8 | 16362965 | 2 | 2.2 | 0.08 |
| LDB_19_44814107 | 19 | 44814107 | 2 | 1.9 | 0.07 |
| LC QTL | 68 | 22 | 61.8 | ||
| SC QTL | 334 | 117 | 36.4 | ||
| 合计Total | 402 | 139 | 98.2 |
表2
基于大豆地方品种资源群体的全基因组关联分析方法比较"
| 性状 Trait | 遗传率 h2 | RTM-GWAS | MLM | ||
|---|---|---|---|---|---|
| QTL | R2 (%) | QTL | R2 (%) | ||
| 油脂含量Oil content | 0.91 | 50 | 82.53 | 3 | 16.69 |
| 油酸含量Oleic acid content | 0.91 | 98 | 90.29 | 18 | 138.76 |
| 亚麻酸含量Linolenic acid content | 0.90 | 50 | 83.34 | 22 | 206.52 |
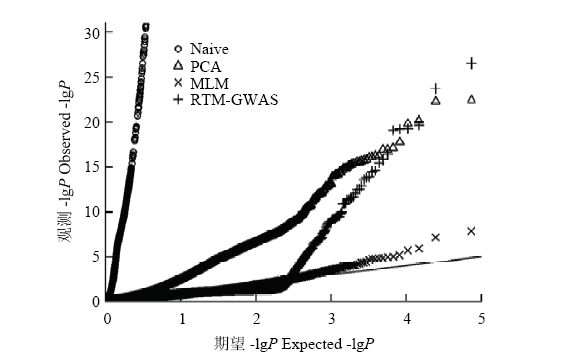
图1
中国大豆种质资源群体百粒重全基因组关联分析Q-Q图 黑色直线为理论分布参考线"
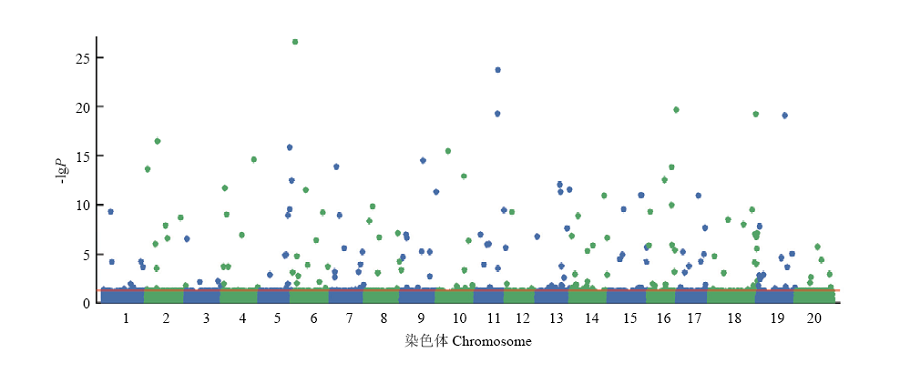
图2
中国大豆种质资源群体百粒重RTM-GWAS分析Manhattan图"
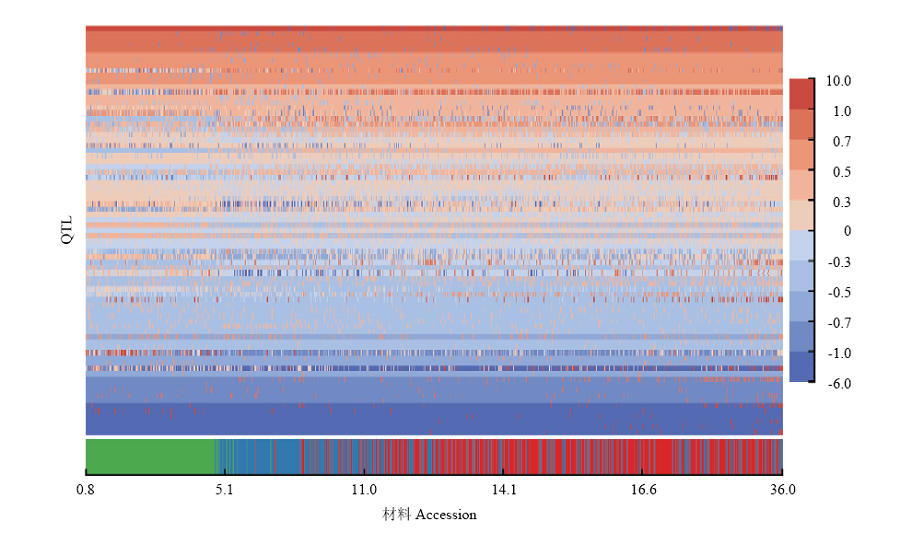
图3
中国大豆种质资源群体百粒重QTL-allele矩阵 横坐标表示材料,按百粒重升序排列,每一列为一个材料的等位基因组成。纵坐标表示QTL,每一行为一个QTL等位基因在材料中的分布。等位基因效应大小使用颜色表示,暖色表示正效,冷色表示负效,颜色深度表示效应大小"
表3
基于大豆NAM群体的五种QTL定位方法特点归纳比较"
| 比较指标 Item | 独立分析 Separate mapping | 联合分析Joint mapping | |||
|---|---|---|---|---|---|
| CIM[ | MCIM[ | JICIM[ | MLM[ | RTM-GWAS | |
| 标记类型 Marker type | BIN | BIN | SNP | SNP | SNPLDB |
| 定位原理 Mapping mechanism | 连锁定位 Linkage mapping | 连锁定位 Linkage mapping | 连锁定位 Linkage mapping | 关联定位 Association mapping | 关联定位 Association mapping |
| QTL数量 Number of QTLs | 8 | 16 | 9 | 7 | 139 |
| 等位基因数量 Number of alleles | 2 | 2 | 8 | 2 | 2~5 |
| 遗传贡献率 Genetic contribution (%) | 73.2—96.1 | 48.4—94.5 | 74.0 | 40.6 | 81.7 |
| 表型数据类型 Phenotype data | 平均数 Entry mean | 小区观测值 Single plot | 平均数 Entry mean | 平均数 Entry mean | 小区观测值 Single plot |
| QTL×环境互作 QTL×Env. | 否No | 是Yes | 否No | 否No | 是Yes |
| 计算机软件 Software | QTL Cartographer | QTLNetwork | QTL IciMapping | TASSEL | RTM-GWAS |
| 命令行界面 Command line | 是Yes | 否No | 否No | 是Yes | 是Yes |
| 计算平台Platform | Windows/Linux | Windows | Windows | Windows/Linux/Mac | Windows/Linux/Mac |
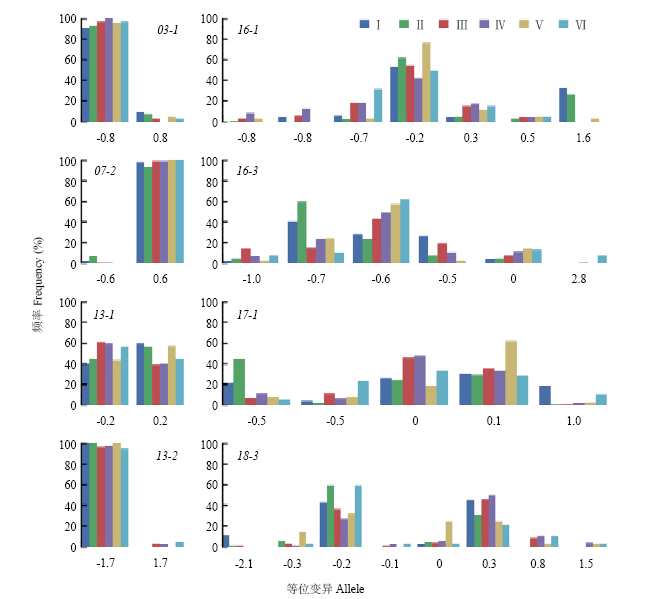
图4
大豆蛋白质含量QTL等位基因在不同生态区的频率分布(张英虎[29]) I:北方一熟制春作生态区;II:黄淮海二熟制春夏作生态区;III:长江中下游二熟制春夏作生态区;IV:中南多熟制春夏秋作生态区;V:西南高原二熟制春夏作生态区;VI:华南热带多熟制四季生态区"
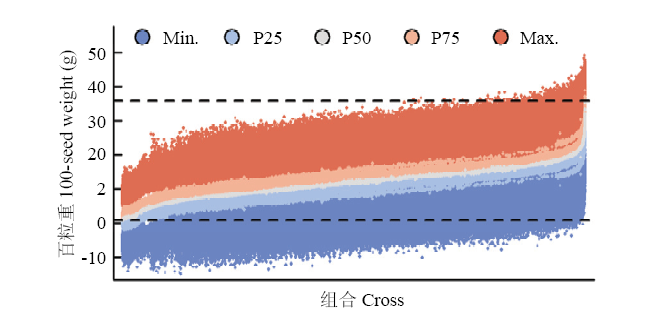
图5
所有单交组合后代预测百粒重分布 两条虚线分别表示亲本观测值的最大值(上)和最小值(下)。Min.、P25、P50、P75和Max.分别表示组合后代预测值的最小值、第25百分位数、第50百分位数、第75百分位数和最大值"
表4
中国大豆种质资源群体百粒重改良优异组合预测"
| 组合 Cross | 观测值 Observation | 99百分位数预测值 99 percentile prediction | |
|---|---|---|---|
| P1 | P2 | ||
| T78205-06×N23548 | 30.4 | 36.0 | 43.1 |
| N23745.0×N23548 | 26.6 | 36.0 | 42.9 |
| N6141×N23548 | 34.0 | 36.0 | 42.4 |
| N04482.1×N23548 | 28.2 | 36.0 | 42.4 |
| N25377×N23548 | 25.5 | 36.0 | 41.6 |
| N23548×N24190 | 36.0 | 26.6 | 41.6 |
| N23548×N05758 | 36.0 | 27.8 | 41.4 |
| N24282×N23548 | 24.8 | 36.0 | 41.4 |
| T78205-06×N05758 | 30.4 | 27.8 | 41.3 |
| N25366×N23548 | 24.4 | 36.0 | 41.2 |
| [1] |
TAM V, PATEL N, TURCOTTE M, BOSSE Y, PARE G, MEYRE D . Benefits and limitations of genome-wide association studies. Nature Reviews Genetics, 2019,20(8):467-484.
doi: 10.1038/s41576-019-0127-1 pmid: 31068683 |
| [2] |
LANDER E S, BOTSTEIN D . Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics, 1989,121(1):185-199.
pmid: 2563713 |
| [3] |
ZENG Z B . Precision mapping of quantitative trait loci. Genetics, 1994,136(4):1457-1468.
pmid: 8013918 |
| [4] |
YU J, HOLLAND J B, MCMULLEN M D, BUCKLER E S . Genetic design and statistical power of nested association mapping in maize. Genetics, 2008,178(1):539-551.
doi: 10.1534/genetics.107.074245 pmid: 18202393 |
| [5] |
MCMULLEN M D, KRESOVICH S, VILLEDA H S, BRADBURY P, LI H, SUN Q, FLINT-GARCIA S, THORNSBERRY J, ACHARYA C, BOTTOMS C, BROWN P, BROWNE C, ELLER M, GUILL K, HARJES C, KROON D, LEPAK N, MITCHELL S E, PETERSON B, PRESSOIR G, ROMERO S, OROPEZA ROSAS M, SALVO S, YATES H, HANSON M, JONES E, SMITH S, GLAUBITZ J C, GOODMAN M, WARE D, HOLLAND J B, BUCKLER E S . Genetic properties of the maize nested association mapping population. Science, 2009,325(5941):737-740.
doi: 10.1126/science.1174320 pmid: 19661427 |
| [6] |
BUCKLER E S, HOLLAND J B, BRADBURY P J, ACHARYA C B, BROWN P J, BROWNE C, ERSOZ E, FLINT-GARCIA S, GARCIA A, GLAUBITZ J C, GOODMAN M M, HARJES C, GUILL K, KROON D E, LARSSON S, LEPAK N K, LI H, MITCHELL S E, PRESSOIR G, PEIFFER J A, ROSAS M O, ROCHEFORD T R, ROMAY M C, ROMERO S, SALVO S, DA SILVA H S, SUN Q, TIAN F, UPADYAYULA N, WARE D, YATES H, YU J, ZHANG Z, KRESOVICH S, MCMULLEN M D . The genetic architecture of maize flowering time. Science, 2009,325(5941):714-718.
doi: 10.1126/science.1174276 pmid: 19661422 |
| [7] |
VISSCHER P M, WRAY N R, ZHANG Q, SKLAR P, MCCARTHY M I, BROWN M A, YANG J . 10 years of GWAS discovery: Biology, function, and translation. American Journal of Human Genetics, 2017,101(1):5-22.
doi: 10.1016/j.ajhg.2017.06.005 pmid: 28686856 |
| [8] |
HUANG X, HAN B . Natural variations and genome-wide association studies in crop plants. Annual Review of Plant Biology, 2014,65:531-551.
doi: 10.1146/annurev-arplant-050213-035715 pmid: 24274033 |
| [9] |
PRICE A L, ZAITLEN N A, REICH D, PATTERSON N . New approaches to population stratification in genome-wide association studies. Nature Reviews Genetics, 2010,11(7):459-463.
doi: 10.1038/nrg2813 pmid: 20548291 |
| [10] |
PRITCHARD J K, STEPHENS M, ROSENBERG N A, DONNELLY P . Association mapping in structured populations. American Journal of Human Genetics, 2000,67(1):170-181.
doi: 10.1086/302959 pmid: 10827107 |
| [11] |
PRICE A L, PATTERSON N J, PLENGE R M, WEINBLATT M E, SHADICK N A, REICH D . Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics, 2006,38(8):904-909.
doi: 10.1038/ng1847 pmid: 16862161 |
| [12] |
YU J, PRESSOIR G, BRIGGS W H, VROH BI I, YAMASAKI M, DOEBLEY J F, MCMULLEN M D, GAUT B S, NIELSEN D M, HOLLAND J B, KRESOVICH S, BUCKLER E S . A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics, 2006,38(2):203-208.
doi: 10.1038/ng1702 pmid: 16380716 |
| [13] |
PRITCHARD J K, STEPHENS M, DONNELLY P . Inference of population structure using multilocus genotype data. Genetics, 2000,155(2):945-959.
pmid: 10835412 |
| [14] |
HE J, MENG S, ZHAO T, XING G, YANG S, LI Y, GUAN R, LU J, WANG Y, XIA Q, YANG B, GAI J . An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theoretical and Applied Genetics, 2017,130(11):2327-2343.
doi: 10.1007/s00122-017-2962-9 pmid: 28828506 |
| [15] |
贺建波, 刘方东, 邢光南, 王吴彬, 赵团结, 管荣展, 盖钧镒 . 限制性两阶段多位点全基因组关联分析方法的特点与计算程序. 作物学报, 2018,44(9):1274-1289.
doi: 10.3724/SP.J.1006.2018.01274 |
|
HE J B, LIU F D, XING G N, WANG W B, ZHAO T J, GUAN R Z, GAI J Y . Characterization and analytical programs of the restricted two-stage multi-locus genome-wide association analysis. Acta Agronomica Sinica, 2018,44(9):1274-1289. (in Chinese)
doi: 10.3724/SP.J.1006.2018.01274 |
|
| [16] |
GABRIEL S B, SCHAFFNER S F, NGUYEN H, MOORE J M, ROY J, BLUMENSTIEL B, HIGGINS J, DEFELICE M, LOCHNER A, FAGGART M, LIU-CORDERO S N, ROTIMI C, ADEYEMO A, COOPER R, WARD R, LANDER E S, DALY M J, ALTSHULER D . The structure of haplotype blocks in the human genome. Science, 2002,296(5576):2225-2229.
doi: 10.1126/science.1069424 pmid: 12029063 |
| [17] |
GAI J, CHEN L, ZHANG Y, ZHAO T, XING G, XING H . Genome-wide genetic dissection of germplasm resources and implications for breeding by design in soybean. Breeding Science, 2012,61(5):495-510.
doi: 10.1270/jsbbs.61.495 pmid: 23136489 |
| [18] |
PATTERSON N, PRICE A L, REICH D . Population structure and eigenanalysis. PLoS Genetics, 2006,2(12):e190.
doi: 10.1371/journal.pgen.0020190 pmid: 17194218 |
| [19] |
VANRADEN P M . Efficient methods to compute genomic predictions. Journal of Dairy Science, 2008,91(11):4414-4423.
doi: 10.3168/jds.2007-0980 pmid: 18946147 |
| [20] |
RISCH N, MERIKANGAS K . The future of genetic studies of complex human diseases. Science, 1996,273(5281):1516-1517.
doi: 10.1126/science.273.5281.1516 pmid: 8801636 |
| [21] |
ZHANG Y, HE J, WANG H, MENG S, XING G, LI Y, YANG S, ZHAO J, ZHAO T, GAI J . Detecting the QTL-allele system of seed oil traits using multi-locus genome-wide association analysis for population characterization and optimal cross prediction in soybean. Frontiers in Plant Science, 2018,9(1793):1793.
doi: 10.3389/fpls.2018.01793 pmid: 30568668 |
| [22] |
PAN L, HE J, ZHAO T, XING G, WANG Y, YU D, CHEN S, GAI J . Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two-stage multi-locus GWAS procedure. Theoretical and Applied Genetics, 2018,131(12):2581-2599.
doi: 10.1007/s00122-018-3174-7 pmid: 30167759 |
| [23] |
LI S, CAO Y, HE J, ZHAO T, GAI J . Detecting the QTL-allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure. Theoretical and Applied Genetics, 2017,130(11):2297-2314.
doi: 10.1007/s00122-017-2960-y pmid: 28799029 |
| [24] |
YANG J, HU C, HU H, YU R, XIA Z, YE X, ZHU J . QTLNetwork: Mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics, 2008,24(5):721-723.
doi: 10.1093/bioinformatics/btm494 pmid: 18202029 |
| [25] | MENG L, LI H H, ZHANG L Y, WANG J K . QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop Journal, 2015,3(3):269-283. |
| [26] |
BRADBURY P J, ZHANG Z, KROON D E, CASSTEVENS T M, RAMDOSS Y, BUCKLER E S . TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics, 2007,23(19):2633-2635.
doi: 10.1093/bioinformatics/btm308 pmid: 17586829 |
| [27] |
KHAN M A, TONG F, WANG W, HE J, ZHAO T, GAI J . Analysis of QTL-allele system conferring drought tolerance at seedling stage in a nested association mapping population of soybean [Glycine max (L.) Merr.] using a novel GWAS procedure. Planta, 2018,248(4):947-962.
doi: 10.1007/s00425-018-2952-4 pmid: 29980855 |
| [28] | ZHANG Y, HE J, MENG S, LIU M, XING G, LI Y, YANG S, YANG J, ZHAO T, GAI J . Identifying QTL-allele system of seed protein content in Chinese soybean landraces for population differentiation studies and optimal cross predictions. Euphytica, 2018,214(9):157. |
| [29] | 张英虎 . 中国大豆地方品种群体籽粒性状的遗传解析及其在设计育种中的应用[D]. 南京: 南京农业大学, 2014. |
| ZHANG Y H . Genetic dissection of seed traits of the Chinese soybean landrace population and its utilization in breeding by design[D]. Nanjing: Nanjing Agricultural University, 2014. (in Chinese) | |
| [30] |
FORSBERG S K, BLOOM J S, SADHU M J, KRUGLYAK L, CARLBORG O . Accounting for genetic interactions improves modeling of individual quantitative trait phenotypes in yeast. Nature Genetics, 2017,49(4):497-503.
doi: 10.1038/ng.3800 pmid: 28250458 |
| [31] |
MACKAY T F . Epistasis and quantitative traits: Using model organisms to study gene-gene interactions. Nature Reviews Genetics, 2014,15(1):22-33.
doi: 10.1038/nrg3627 pmid: 24296533 |
| [32] |
WEI W H, HEMANI G, HALEY C S . Detecting epistasis in human complex traits. Nature Reviews Genetics, 2014,15(11):722-733.
doi: 10.1038/nrg3747 pmid: 25200660 |
| [33] |
WAN X, YANG C, YANG Q, XUE H, FAN X, TANG N L, YU W . BOOST: A fast approach to detecting gene-gene interactions in genome-wide case-control studies. American Journal of Human Genetics, 2010,87(3):325-340.
doi: 10.1016/j.ajhg.2010.07.021 pmid: 20817139 |
| [34] |
ZHANG X, HUANG S, ZOU F, WANG W . TEAM: Efficient two-locus epistasis tests in human genome-wide association study. Bioinformatics, 2010,26(12):i217-i227.
doi: 10.1093/bioinformatics/btq186 pmid: 20529910 |
| [35] |
SCHADT E E, LINDERMAN M D, SORENSON J, LEE L, NOLAN G P . Computational solutions to large-scale data management and analysis. Nature Reviews Genetics, 2010,11(9):647-657.
doi: 10.1038/nrg2857 pmid: 20717155 |
| [36] |
ZHANG F T, ZHU Z H, TONG X R, ZHU Z X, QI T, ZHU J . Mixed linear model approaches of association mapping for complex traits based on omics variants. Scientific Reports, 2015,5:10298.
doi: 10.1038/srep10298 pmid: 26223539 |
| [37] |
MEUWISSEN T H, HAYES B J, GODDARD M E . Prediction of total genetic value using genome-wide dense marker maps. Genetics, 2001,157(4):1819-1829.
pmid: 11290733 |
| [1] | 叶美金, 吴雷, Lohani Md Nahibuzzaman, 尹丽, 胡欣荣, 刘亚西, 蒋云峰, 陈国跃, 蒲至恩, 李阳, 李婷, 邹亚亚, 吴佳怡, 马建. 基于GWAS的中国地方小麦成熟胚大小位点的鉴定及其遗传效应解析[J]. 中国农业科学, 2026, 59(6): 1157-1171. |
| [2] | 杨丽娟, 陈丝雨, 赵薇, 朱玲, 郭磊, 马丽娜, 马瑞敏, 张娟. 全基因组重测序揭示静原鸡羽色的遗传机制[J]. 中国农业科学, 2026, 59(6): 1348-1360. |
| [3] | 王勇胜, 牛丽, 王长杰, 马立花, 廉潇潇, 孟亚雄, 马小乐, 姚立蓉, 张宏, 杨轲, 李葆春, 王化俊, 司二静, 汪军成. 冬小麦千粒重的全基因组关联分析及候选基因预测[J]. 中国农业科学, 2026, 59(3): 499-514. |
| [4] | 李云丽, 刁邓超, 刘雅睿, 孙玉晨, 孟祥宇, 邬陈芳, 汪妤, 吴建辉, 李春莲, 曾庆东, 韩德俊, 郑炜君. 小麦苗期耐热性全基因组关联分析[J]. 中国农业科学, 2025, 58(9): 1663-1683. |
| [5] | 周广飞, 马亮, 马璐, 张舒钰, 章慧敏, 宋旭东, 张振良, 陆虎华, 郝德荣, 冒宇翔, 薛林, 陈国清. 玉米苞叶性状全基因组关联分析[J]. 中国农业科学, 2025, 58(3): 431-442. |
| [6] | 武书羽, 衡燕芳, 于太飞, 王世佳, 于思佳, 李园, 胡正, 张辉, 孙现军, 黎亮, 姜奇彦. 玉米自然群体苗期耐盐性鉴定及耐盐相关基因分析[J]. 中国农业科学, 2025, 58(20): 4085-4099. |
| [7] | 向爱慧, 白荣基, 郝宇琼, 赵佳佳, 武棒棒, 李晓华, 郑兴卫, 关攀锋, 郑军. 山西小麦矮秆基因的鉴定及株高遗传位点挖掘[J]. 中国农业科学, 2025, 58(17): 3372-3388. |
| [8] | 郑敏华, 陈洛, 邢甲乐, 谢月兰, 姜先芽, 聂帅, 蔡甫格, 巫浩翔, 陆展华, 孙伟, 霍兴, 白嵩, 赵均良, 杨武. 华南籼稻稻瘟病抗性QTL鉴定与候选基因挖掘[J]. 中国农业科学, 2025, 58(14): 2707-2719. |
| [9] | 李宁, 高丽锋, 黄鑫, 史华伟, 杨进文, 史雨刚, 陈明, 贾继增, 孙黛珍. 耐低氮小麦品种的筛选及耐低氮指数的全基因组关联分析[J]. 中国农业科学, 2025, 58(13): 2487-2503. |
| [10] | 史顺宇, 杨涛, 庞博, 李静, 林轶峰, 王正瑞, 傅林成, 扎尔加玛丽·阿不都别克, 高文伟, 吴鹏昊. 海岛棉叶绿素含量的全基因组关联分析及候选基因预测[J]. 中国农业科学, 2025, 58(10): 1878-1895. |
| [11] | 张颖, 石婷瑞, 曹瑞, 潘文秋, 宋卫宁, 王利, 聂小军. ICARDA引进-小麦苗期抗旱性的全基因组关联分析[J]. 中国农业科学, 2024, 57(9): 1658-1673. |
| [12] | 赵真坚, 王凯, 陈栋, 申琦, 余杨, 崔晟頔, 王俊戈, 陈子旸, 禹世欣, 陈佳苗, 王翔枫, 唐国庆. 基因组和DNA甲基化组联合分析筛选猪肉质性状关键基因[J]. 中国农业科学, 2024, 57(7): 1394-1406. |
| [13] | 郭军, 邵丹, 窦套存, 马猛, 卢建, 胡玉萍, 王星果, 王强, 李永峰, 郭伟, 童海兵, 曲亮. 鸡产蛋期剩余采食量的随机回归分析及遗传标记筛选[J]. 中国农业科学, 2024, 57(22): 4568-4577. |
| [14] | 郭军, 曲亮, 邵丹, 马猛, 窦套存, 卢建, 胡玉萍, 王星果, 王强, 李永峰, 郭伟, 童海兵. 基于一步法全基因组关联分析解析蛋黄比率遗传结构[J]. 中国农业科学, 2024, 57(21): 4356-4366. |
| [15] | 白冰楠, 乔丹, 葛群, 栾玉娟, 刘小芳, 卢全伟, 牛皓, 龚举武, 巩万奎, ELAMEER ELSAMMAN, 闫浩亮, 李俊文, 刘爱英, 石玉真, 王海泽, 袁有禄. 陆地棉棉籽相关性状的QTN挖掘及候选基因筛选[J]. 中国农业科学, 2024, 57(15): 2901-2913. |
|
||