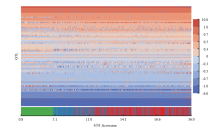
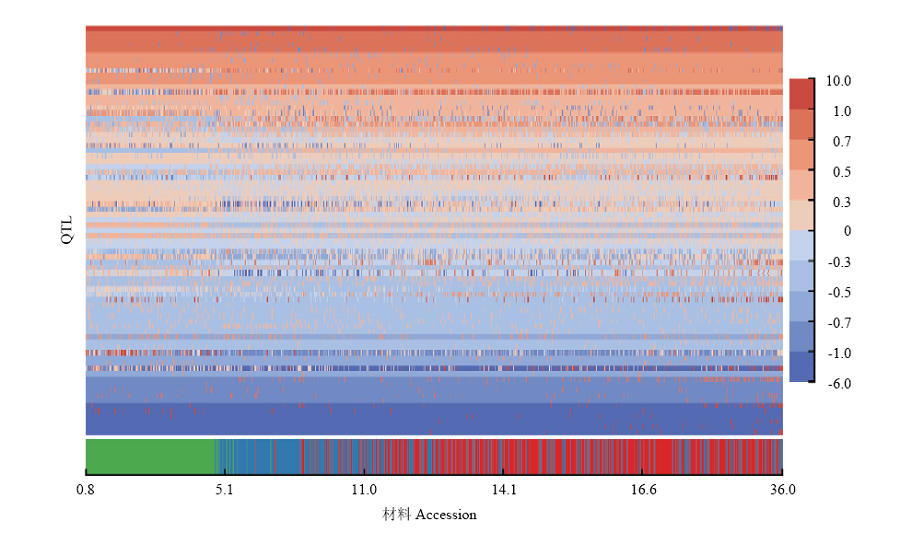
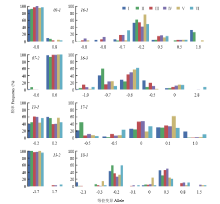
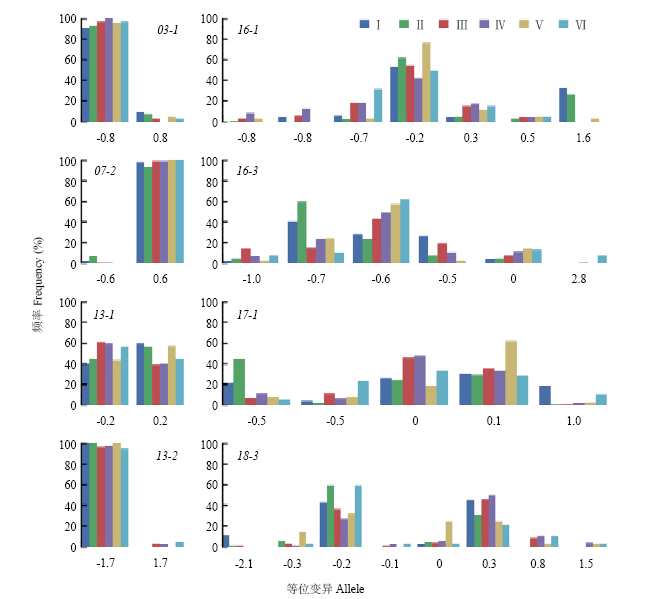
Scientia Agricultura Sinica ›› 2020, Vol. 53 ›› Issue (9): 1704-1716.doi: 10.3864/j.issn.0578-1752.2020.09.002
• SPECIAL FOCUS: APPLICATIONS OF RESTRICTED TWO-STAGE MULTI-LOCUS GENOME-WIDE ASSOCIATION ANALYSIS • Previous Articles Next Articles
JianBo HE,FangDong LIU,WuBin WANG,GuangNan XING,RongZhan GUAN,JunYi GAI( )
)
| [1] |
TAM V, PATEL N, TURCOTTE M, BOSSE Y, PARE G, MEYRE D . Benefits and limitations of genome-wide association studies. Nature Reviews Genetics, 2019,20(8):467-484.
doi: 10.1038/s41576-019-0127-1 pmid: 31068683 |
| [2] |
LANDER E S, BOTSTEIN D . Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics, 1989,121(1):185-199.
pmid: 2563713 |
| [3] |
ZENG Z B . Precision mapping of quantitative trait loci. Genetics, 1994,136(4):1457-1468.
pmid: 8013918 |
| [4] |
YU J, HOLLAND J B, MCMULLEN M D, BUCKLER E S . Genetic design and statistical power of nested association mapping in maize. Genetics, 2008,178(1):539-551.
doi: 10.1534/genetics.107.074245 pmid: 18202393 |
| [5] |
MCMULLEN M D, KRESOVICH S, VILLEDA H S, BRADBURY P, LI H, SUN Q, FLINT-GARCIA S, THORNSBERRY J, ACHARYA C, BOTTOMS C, BROWN P, BROWNE C, ELLER M, GUILL K, HARJES C, KROON D, LEPAK N, MITCHELL S E, PETERSON B, PRESSOIR G, ROMERO S, OROPEZA ROSAS M, SALVO S, YATES H, HANSON M, JONES E, SMITH S, GLAUBITZ J C, GOODMAN M, WARE D, HOLLAND J B, BUCKLER E S . Genetic properties of the maize nested association mapping population. Science, 2009,325(5941):737-740.
doi: 10.1126/science.1174320 pmid: 19661427 |
| [6] |
BUCKLER E S, HOLLAND J B, BRADBURY P J, ACHARYA C B, BROWN P J, BROWNE C, ERSOZ E, FLINT-GARCIA S, GARCIA A, GLAUBITZ J C, GOODMAN M M, HARJES C, GUILL K, KROON D E, LARSSON S, LEPAK N K, LI H, MITCHELL S E, PRESSOIR G, PEIFFER J A, ROSAS M O, ROCHEFORD T R, ROMAY M C, ROMERO S, SALVO S, DA SILVA H S, SUN Q, TIAN F, UPADYAYULA N, WARE D, YATES H, YU J, ZHANG Z, KRESOVICH S, MCMULLEN M D . The genetic architecture of maize flowering time. Science, 2009,325(5941):714-718.
doi: 10.1126/science.1174276 pmid: 19661422 |
| [7] |
VISSCHER P M, WRAY N R, ZHANG Q, SKLAR P, MCCARTHY M I, BROWN M A, YANG J . 10 years of GWAS discovery: Biology, function, and translation. American Journal of Human Genetics, 2017,101(1):5-22.
doi: 10.1016/j.ajhg.2017.06.005 pmid: 28686856 |
| [8] |
HUANG X, HAN B . Natural variations and genome-wide association studies in crop plants. Annual Review of Plant Biology, 2014,65:531-551.
doi: 10.1146/annurev-arplant-050213-035715 pmid: 24274033 |
| [9] |
PRICE A L, ZAITLEN N A, REICH D, PATTERSON N . New approaches to population stratification in genome-wide association studies. Nature Reviews Genetics, 2010,11(7):459-463.
doi: 10.1038/nrg2813 pmid: 20548291 |
| [10] |
PRITCHARD J K, STEPHENS M, ROSENBERG N A, DONNELLY P . Association mapping in structured populations. American Journal of Human Genetics, 2000,67(1):170-181.
doi: 10.1086/302959 pmid: 10827107 |
| [11] |
PRICE A L, PATTERSON N J, PLENGE R M, WEINBLATT M E, SHADICK N A, REICH D . Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics, 2006,38(8):904-909.
doi: 10.1038/ng1847 pmid: 16862161 |
| [12] |
YU J, PRESSOIR G, BRIGGS W H, VROH BI I, YAMASAKI M, DOEBLEY J F, MCMULLEN M D, GAUT B S, NIELSEN D M, HOLLAND J B, KRESOVICH S, BUCKLER E S . A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics, 2006,38(2):203-208.
doi: 10.1038/ng1702 pmid: 16380716 |
| [13] |
PRITCHARD J K, STEPHENS M, DONNELLY P . Inference of population structure using multilocus genotype data. Genetics, 2000,155(2):945-959.
pmid: 10835412 |
| [14] |
HE J, MENG S, ZHAO T, XING G, YANG S, LI Y, GUAN R, LU J, WANG Y, XIA Q, YANG B, GAI J . An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theoretical and Applied Genetics, 2017,130(11):2327-2343.
doi: 10.1007/s00122-017-2962-9 pmid: 28828506 |
| [15] |
贺建波, 刘方东, 邢光南, 王吴彬, 赵团结, 管荣展, 盖钧镒 . 限制性两阶段多位点全基因组关联分析方法的特点与计算程序. 作物学报, 2018,44(9):1274-1289.
doi: 10.3724/SP.J.1006.2018.01274 |
|
HE J B, LIU F D, XING G N, WANG W B, ZHAO T J, GUAN R Z, GAI J Y . Characterization and analytical programs of the restricted two-stage multi-locus genome-wide association analysis. Acta Agronomica Sinica, 2018,44(9):1274-1289. (in Chinese)
doi: 10.3724/SP.J.1006.2018.01274 |
|
| [16] |
GABRIEL S B, SCHAFFNER S F, NGUYEN H, MOORE J M, ROY J, BLUMENSTIEL B, HIGGINS J, DEFELICE M, LOCHNER A, FAGGART M, LIU-CORDERO S N, ROTIMI C, ADEYEMO A, COOPER R, WARD R, LANDER E S, DALY M J, ALTSHULER D . The structure of haplotype blocks in the human genome. Science, 2002,296(5576):2225-2229.
doi: 10.1126/science.1069424 pmid: 12029063 |
| [17] |
GAI J, CHEN L, ZHANG Y, ZHAO T, XING G, XING H . Genome-wide genetic dissection of germplasm resources and implications for breeding by design in soybean. Breeding Science, 2012,61(5):495-510.
doi: 10.1270/jsbbs.61.495 pmid: 23136489 |
| [18] |
PATTERSON N, PRICE A L, REICH D . Population structure and eigenanalysis. PLoS Genetics, 2006,2(12):e190.
doi: 10.1371/journal.pgen.0020190 pmid: 17194218 |
| [19] |
VANRADEN P M . Efficient methods to compute genomic predictions. Journal of Dairy Science, 2008,91(11):4414-4423.
doi: 10.3168/jds.2007-0980 pmid: 18946147 |
| [20] |
RISCH N, MERIKANGAS K . The future of genetic studies of complex human diseases. Science, 1996,273(5281):1516-1517.
doi: 10.1126/science.273.5281.1516 pmid: 8801636 |
| [21] |
ZHANG Y, HE J, WANG H, MENG S, XING G, LI Y, YANG S, ZHAO J, ZHAO T, GAI J . Detecting the QTL-allele system of seed oil traits using multi-locus genome-wide association analysis for population characterization and optimal cross prediction in soybean. Frontiers in Plant Science, 2018,9(1793):1793.
doi: 10.3389/fpls.2018.01793 pmid: 30568668 |
| [22] |
PAN L, HE J, ZHAO T, XING G, WANG Y, YU D, CHEN S, GAI J . Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two-stage multi-locus GWAS procedure. Theoretical and Applied Genetics, 2018,131(12):2581-2599.
doi: 10.1007/s00122-018-3174-7 pmid: 30167759 |
| [23] |
LI S, CAO Y, HE J, ZHAO T, GAI J . Detecting the QTL-allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure. Theoretical and Applied Genetics, 2017,130(11):2297-2314.
doi: 10.1007/s00122-017-2960-y pmid: 28799029 |
| [24] |
YANG J, HU C, HU H, YU R, XIA Z, YE X, ZHU J . QTLNetwork: Mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics, 2008,24(5):721-723.
doi: 10.1093/bioinformatics/btm494 pmid: 18202029 |
| [25] | MENG L, LI H H, ZHANG L Y, WANG J K . QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop Journal, 2015,3(3):269-283. |
| [26] |
BRADBURY P J, ZHANG Z, KROON D E, CASSTEVENS T M, RAMDOSS Y, BUCKLER E S . TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics, 2007,23(19):2633-2635.
doi: 10.1093/bioinformatics/btm308 pmid: 17586829 |
| [27] |
KHAN M A, TONG F, WANG W, HE J, ZHAO T, GAI J . Analysis of QTL-allele system conferring drought tolerance at seedling stage in a nested association mapping population of soybean [Glycine max (L.) Merr.] using a novel GWAS procedure. Planta, 2018,248(4):947-962.
doi: 10.1007/s00425-018-2952-4 pmid: 29980855 |
| [28] | ZHANG Y, HE J, MENG S, LIU M, XING G, LI Y, YANG S, YANG J, ZHAO T, GAI J . Identifying QTL-allele system of seed protein content in Chinese soybean landraces for population differentiation studies and optimal cross predictions. Euphytica, 2018,214(9):157. |
| [29] | 张英虎 . 中国大豆地方品种群体籽粒性状的遗传解析及其在设计育种中的应用[D]. 南京: 南京农业大学, 2014. |
| ZHANG Y H . Genetic dissection of seed traits of the Chinese soybean landrace population and its utilization in breeding by design[D]. Nanjing: Nanjing Agricultural University, 2014. (in Chinese) | |
| [30] |
FORSBERG S K, BLOOM J S, SADHU M J, KRUGLYAK L, CARLBORG O . Accounting for genetic interactions improves modeling of individual quantitative trait phenotypes in yeast. Nature Genetics, 2017,49(4):497-503.
doi: 10.1038/ng.3800 pmid: 28250458 |
| [31] |
MACKAY T F . Epistasis and quantitative traits: Using model organisms to study gene-gene interactions. Nature Reviews Genetics, 2014,15(1):22-33.
doi: 10.1038/nrg3627 pmid: 24296533 |
| [32] |
WEI W H, HEMANI G, HALEY C S . Detecting epistasis in human complex traits. Nature Reviews Genetics, 2014,15(11):722-733.
doi: 10.1038/nrg3747 pmid: 25200660 |
| [33] |
WAN X, YANG C, YANG Q, XUE H, FAN X, TANG N L, YU W . BOOST: A fast approach to detecting gene-gene interactions in genome-wide case-control studies. American Journal of Human Genetics, 2010,87(3):325-340.
doi: 10.1016/j.ajhg.2010.07.021 pmid: 20817139 |
| [34] |
ZHANG X, HUANG S, ZOU F, WANG W . TEAM: Efficient two-locus epistasis tests in human genome-wide association study. Bioinformatics, 2010,26(12):i217-i227.
doi: 10.1093/bioinformatics/btq186 pmid: 20529910 |
| [35] |
SCHADT E E, LINDERMAN M D, SORENSON J, LEE L, NOLAN G P . Computational solutions to large-scale data management and analysis. Nature Reviews Genetics, 2010,11(9):647-657.
doi: 10.1038/nrg2857 pmid: 20717155 |
| [36] |
ZHANG F T, ZHU Z H, TONG X R, ZHU Z X, QI T, ZHU J . Mixed linear model approaches of association mapping for complex traits based on omics variants. Scientific Reports, 2015,5:10298.
doi: 10.1038/srep10298 pmid: 26223539 |
| [37] |
MEUWISSEN T H, HAYES B J, GODDARD M E . Prediction of total genetic value using genome-wide dense marker maps. Genetics, 2001,157(4):1819-1829.
pmid: 11290733 |
| [1] | JunYi GAI,JianBo HE. Major Characteristics, Often-Raised Queries and Potential Usefulness of the Restricted Two-Stage Multi-Locus Genome-Wide Association Analysis [J]. Scientia Agricultura Sinica, 2020, 53(9): 1699-1703. |
| [2] | XiaoShuai HAO,MengMeng FU,ZaiDong LIU,JianBo HE,YanPing WANG,HaiXiang REN,DeLiang WANG,XingYong YANG,YanXi CHENG,WeiGuang DU,JunYi GAI. Genome-Wide QTL-Allele Dissection of 100-Seed Weight in the Northeast China Soybean Germplasm Population [J]. Scientia Agricultura Sinica, 2020, 53(9): 1717-1729. |
| [3] | LiYuan PAN,JianBo HE,JinMing ZHAO,WuBin WANG,GuangNan XING,DeYue YU,XiaoYan ZHANG,ChunYan LI,ShouYi CHEN,JunYi GAI. Detection Power of RTM-GWAS Applied to 100-Seed Weight QTL Identification in a Recombinant Inbred Lines Population of Soybean [J]. Scientia Agricultura Sinica, 2020, 53(9): 1730-1742. |
| [4] | ShuGuang LI,YongCe CAO,JianBo HE,WuBin WANG,GuangNan XING,JiaYin YANG,TuanJie ZHAO,JunYi GAI. Genetic Dissection of Protein Content in a Nested Association Mapping Population of Soybean [J]. Scientia Agricultura Sinica, 2020, 53(9): 1743-1755. |
| [5] | ZHANG Ying-hu, MENG Shan, HE Jian-bo, WANG Yu-feng, XING Guang-nan, ZHAO Tuan-jie, GAI Jun-yi. The Genetic Constitution of Transgressive Segregation of the 100-Seed Weight in A Recombinant Inbred Line Population NJRSXG of Soybean [J]. Scientia Agricultura Sinica, 2015, 48(22): 4408-4416. |
| [6] | ,. Studies on Inheritance of Symptom Reaction to Soybean Mosaic Virus in Soybean [J]. Scientia Agricultura Sinica, 2005, 38(05): 944-949 . |
| [7] | . Studies of Multiple-Allelic Polymorphism of Dominant Dwarfing Genes in Wheat [J]. Scientia Agricultura Sinica, 2004, 37(09): 1251-1260 . |
|
||