






中国农业科学 ›› 2020, Vol. 53 ›› Issue (4): 683-694.doi: 10.3864/j.issn.0578-1752.2020.04.002
曹永策1,2,李曙光2,张新草1,孔杰杰2,赵团结2( )
)
收稿日期:2019-07-31
接受日期:2019-09-29
出版日期:2020-02-16
发布日期:2020-03-09
联系方式:
曹永策,E-mail:caoyongce@yau.edu.cn。
基金资助:
YongCe CAO1,2,ShuGuang LI2,XinCao ZHANG1,JieJie KONG2,TuanJie ZHAO2()
Received:2019-07-31
Accepted:2019-09-29
Published:2020-02-16
Online:2020-03-09
摘要:
【背景】开花期是大豆重要的生育期性状,不仅决定了大豆品种的适种范围,而且对大豆的产量和品质有重要影响。江淮地区是中国重要的大豆产区,目前对该地区夏大豆开花期性状遗传基础研究相对较少。【目的】利用2个夏大豆材料杂交衍生的重组自交系群体对开花期进行QTL定位,为分子标记辅助选择育种和基因克隆提供依据。【方法】以科丰35(KF35)和南农1138-2(NN1138-2)为亲本,构建了含91个家系(F2:8)的重组自交系群体(NJK3N-RIL),在6个环境下调查开花期性状数据。利用限制位点相关DNA测序(restriction-site associated DNA sequencing,RAD-seq)技术对群体亲本及家系材料进行SNP标记分型,并利用窗口滑动法进行bin标记划分。利用bin标记构建该群体的遗传图谱,结合多年多点的表型数据,使用QTL Network 2.2软件中的基于混合线性模型的复合区间作图法(mixed-model based composite interval mapping,MCIM)和Windows QTL Cartographer V2.5_011软件中的复合区间作图法(composite interval mapping,CIM)对开花期性状进行QTL分析。【结果】在大豆全基因组范围内共获得36 778个高质量SNP标记,被划分为1 733个bin标记。利用1 733个bin标记构建了一张覆盖大豆20条染色体遗传图谱,图谱长度为2 362.4 cM,标记间平均遗传距离为1.4 cM。利用MCIM法共检测到9个控制开花期的加性QTL、2对上位性QTL和1个环境互作QTL,3种效应累积贡献率分别为63.9%、4.6%和2.1%。利用CIM法共检测到10个控制开花期的QTL,其中qFT-8-1、qFT-11-1、qFT-15-1、qFT-16-1能在3个及以上环境检测到。综合2种分析方法,共检测到12个开花期QTL,其中qFT-8-1、qFT-11-1、qFT-15-1、qFT-16-1、qFT-16-2、qFT-20-1和qFT-20-2等能够被2种方法检测到。同时qFT-5-1、qFT-8-1、qFT-8-2、qFT-13-1、qFT-15-1和qFT-20-2等是本研究新检测到的开花期QTL。【结论】夏大豆开花期遗传构成复杂,但加性QTL效应占绝对优势,上位性互作及环境互作效应对开花期影响较小。qFT-8-1、qFT-11-1、qFT-15-1、qFT-16-1能够被2种方法在多个环境中检测到,是NJK3N-RIL群体中控制开花期的重要位点。
曹永策, 李曙光, 张新草, 孔杰杰, 赵团结. 夏大豆重组自交系群体遗传图谱构建及开花期QTL分析[J]. 中国农业科学, 2020, 53(4): 683-694.
YongCe CAO, ShuGuang LI, XinCao ZHANG, JieJie KONG, TuanJie ZHAO. Construction of Genetic Map and Mapping QTL for Flowering Time in A Summer Planting Soybean Recombinant Inbred Line Population[J]. Scientia Agricultura Sinica, 2020, 53(4): 683-694.
表1
不同环境下NJK3N-RIL群体及其亲本开花期(天)性状描述性统计"
| 环境 Environment | 亲本 Parents | NJK3N-RIL群体 | ||||||
|---|---|---|---|---|---|---|---|---|
| 科丰35 KF35 (d) | 南农1138-2 NN1138-2 (d) | 均值±标准差 Mean ± SD (d) | 变幅 Range (d) | 偏度 Skewness | 峰度 Kurtosis | 变异系数 CV (%) | ||
| 2012JP | 36.8 ± 2.4 | 39.8 ± 1.5 | 37.7 ± 1.9 | 31.0—41.3 | -0.5 | 0.3 | 5.2 | |
| 2012FY | 48.3 ± 2.3 | 54.0 ± 4.6 | 51.1 ± 2.0 | 47.7—57.3 | 0.3 | -0.3 | 4.0 | |
| 2013JP | 40.7 ± 3.1 | 50.5 ± 3.7 | 44.8 ± 2.4 | 40.0—50.7 | 0.3 | -0.4 | 5.3 | |
| 2013FY | 41.0± 0.0 | 51.3 ± 2.0 | 47.1 ± 2.5 | 43.0—53.7 | 0.9 | 0.6 | 5.4 | |
| 2014JP | 40.3 ± 2.3 | 47.0 ± 1.3 | 49.3 ± 1.9 | 37.7—48.7 | -0.4 | 1.0 | 4.2 | |
| 2014YC | 35.3 ± 0.6 | 45.7 ± 1.2 | 42.1 ± 2.6 | 35.7—48.3 | 0.4 | 0.1 | 6.2 | |

图1
NJK3N-RIL群体开花期在不同环境中的分布"
表2
NJK3N-RIL群体遗传图谱信息汇总"
| 染色体 Chromosome | SNP数目 SNP numbers | bin标记数目 bin numbers | 遗传图谱长度 Linkage distance (cM) | 标记间平均距离 Mean distance of adjacent markers(cM) |
|---|---|---|---|---|
| Chr. 01 | 528 | 57 | 110.3 | 1.9 |
| Chr. 02 | 1127 | 114 | 146.4 | 1.3 |
| Chr. 03 | 3521 | 81 | 110.2 | 1.4 |
| Chr. 04 | 1272 | 90 | 126.4 | 1.4 |
| Chr. 05 | 1333 | 73 | 94.5 | 1.3 |
| Chr. 06 | 1497 | 87 | 133.6 | 1.5 |
| Chr. 07 | 1700 | 85 | 132.1 | 1.6 |
| Chr. 08 | 1111 | 109 | 144.5 | 1.3 |
| Chr. 09 | 1030 | 85 | 140.7 | 1.7 |
| Chr. 10 | 3435 | 95 | 123.9 | 1.3 |
| Chr. 11 | 2256 | 85 | 126.4 | 1.5 |
| Chr. 12 | 733 | 65 | 115.5 | 1.8 |
| Chr. 13 | 2264 | 104 | 125.5 | 1.2 |
| Chr. 14 | 1752 | 81 | 80.1 | 1.0 |
| Chr. 15 | 2008 | 88 | 148.6 | 1.7 |
| Chr. 16 | 2384 | 84 | 85.4 | 1.0 |
| Chr. 17 | 2220 | 84 | 118.0 | 1.4 |
| Chr. 18 | 4513 | 110 | 98.2 | 0.9 |
| Chr. 19 | 695 | 71 | 96.8 | 1.4 |
| Chr. 20 | 1399 | 85 | 105.5 | 1.2 |
| 总计Total | 36778 | 1733 | 2362.4 | 1.4 |
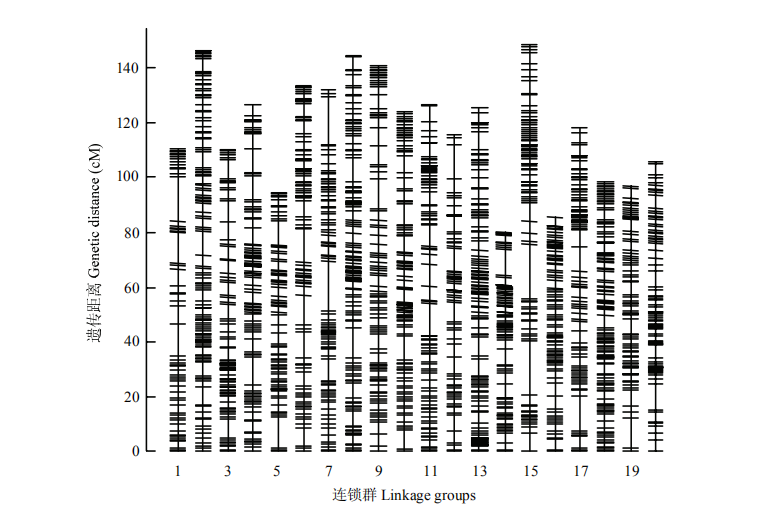
图2
NJK3N-RIL群体中bin标记在遗传图谱上的分布"
表3
NJK3N-RIL群体开花期加性及QTL与环境互作效应的分析"
| 位点 QTL | 染色体 Chromosome | 两侧标记 Flank markers | 位置 Position (cM) | QTL置信区间 Confidence interval (cM) | 物理区间 1-LOD interval (Mb) | 加性效应 A (d) | h2A (%) | 加性与环境 互作效应 AE (d) | h2AE (%) | 报道位点 Reported locus |
|---|---|---|---|---|---|---|---|---|---|---|
| qFT-8-1 | 8 | bin670-bin671 | 98.2 | 95.6—102.2 | 36.6—41.3 | 0.4 | 3.2 | 新位点 Novel | ||
| qFT-10-1 | 10 | bin875-bin876 | 123.3 | 122.0—123.3 | 48.6—49.3 | -0.5 | 4.8 | First flower 24-4 | ||
| qFT-11-1 | 11 | bin928-bin929 | 85.8 | 84.6—86.5 | 14.9—15.6 | 0.7 | 9.9 | 0.6**(2013FY) -0.8**(2014JP) | 4.6 | First flower 11-2 |
| qFT-13-1 | 13 | bin1093-bin1094 | 68.0 | 66.8—68.5 | 28.4—29.5 | 0.4 | 3.0 | 新位点 Novel | ||
| qFT-15-1 | 15 | bin1289-bin1290 | 130.1 | 128.1—130.1 | 48.1—49.0 | 0.6 | 7.9 | 新位点 Novel | ||
| qFT-16-1 | 16 | bin1313-bin1314 | 24.2 | 21.2—27.7 | 3.8—5.0 | 0.7 | 10.5 | First flower 13-7 | ||
| qFT-16-2 | 16 | bin1357-bin1358 | 64.3 | 62.2—66.2 | 31.1—32.0 | 0.6 | 7.2 | First flower 9-3 | ||
| qFT-20-1 | 20 | bin1688-bin1689 | 47.7 | 46.8—48.3 | 34.3—34.5 | 0.7 | 12.0 | First flower 21-2 | ||
| qFT-20-2 | 20 | bin1727-bin1728 | 95.8 | 95.2—96.8 | 44.0—44.5 | 0.5 | 5.4 | 新位点 Novel |

图3
NJK3N-RIL群体中开花期QTL在连锁群上的位置 方条的长度代表QTL置信区间。空白长条代表使用CIM方法在不同环境中定位到的QTL;黑色长条代表使用MCIM在联合环境中定位到的QTL;虚线代表相连的QTL间具有上位性互作"
表4
NJK3N-RIL群体开花期QTL上位互作效应分析"
| 上位性QTL对 Epistatic QTL pairs | 位点 QTL | 相邻标记 Flanking marker | 位置 position (cM) | 置信区间 Confidence interval (cM) | 上位性效应 AA (d) | h2AA (%) | 上位性与环境互作效应 AAE (d) | h2AAE (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | qFT-11-1 | bin928-bin929 | 85.8 | 84.6—86.5 | -0.2 | 0.6 | -0.3**(2013FY) 0.2*(2014JP) | 0.8 |
| qFT-16-1 | bin1313-bin1314 | 24.2 | 21.2—27.7 | |||||
| 2 | qFT-13-1 | bin1093-bin1094 | 68.0 | 66.8—68.5 | 0.3 | 1.5 | ||
| qFT-16-2 | bin1357-bin1358 | 64.3 | 62.2—66.2 |
表5
NJK3N-RIL群体不同环境中开花期QTL分析"
| 位点 QTL | 染色体 Chromosome | 遗传位置 Position (cM) | 相邻标记 Flank markers | LOD | 置信区间 Confidence interval (cM) | 加性效应 Additive (d) | 贡献率 R2 (%) | 环境 Environment | 报道位点 Reported locus |
|---|---|---|---|---|---|---|---|---|---|
| qFT-5-1 | 5 | 69.4 | bin398-bin399 | 4.4 | 67.5—71.8 | -0.6 | 9.9 | 2014JP | Novel |
| qFT-6-1 | 6 | 108.3 | bin482-bin483 | 3.8 | 106.9—109.5 | 0.6 | 5.0 | 2013JP | First flower 1-1 |
| qFT-8-1 | 8 | 105.2 | bin670-bin671 | 4.5 | 99.2—108.2 | 0.8 | 9.1 | 2013FY | Novel |
| 107.2 | bin671-bin672 | 3.2 | 102.9—108.4 | 0.5 | 6.6 | 2012JP | |||
| 106.2 | bin670-bin671 | 4.6 | 100.9—111.3 | 0.6 | 9.0 | 2014YC | |||
| qFT-8-2 | 8 | 123.1 | bin685-bin686 | 4.8 | 121.4—125.2 | 0.7 | 9.4 | 2012FY | Novel |
| 123.1 | bin685-bin686 | 3.8 | 121.3—125.2 | 0.6 | 5.9 | 2013JP | |||
| qFT-11-1 | 11 | 84.6 | bin927-bin928 | 12.7 | 84.2—85.8 | 1.3 | 34.3 | 2012JP | First flower 11-2 |
| 84.5 | bin927-bin928 | 14.2 | 83.6—87.5 | 1.5 | 39.6 | 2012FY | |||
| 87.2 | bin930-bin931 | 18.2 | 86.5—88.4 | 1.9 | 49.6 | 2013FY | |||
| 88.2 | bin930-bin931 | 8.6 | 86.8—89.2 | 1.0 | 18.5 | 2014YC | |||
| 92.9 | bin932-bin933 | 9.4 | 89.0—95.6 | 1.1 | 17.7 | 2013JP | |||
| qFT-15-1 | 15 | 130.1 | bin1290-bin1291 | 9.3 | 127.4—133.5 | 0.9 | 23.0 | 2014JP | Novel |
| 134.6 | bin1291-bin1292 | 4.5 | 130.6—136.8 | 0.7 | 7.6 | 2013JP | |||
| 134.6 | bin1291-bin1292 | 4.6 | 131.0—138.3 | 0.6 | 8.3 | 2014YC | |||
| 135.4 | bin1292-bin1293 | 4.0 | 130.6—138.8 | 0.6 | 8.6 | 2012JP | |||
| qFT-16-1 | 16 | 21.3 | bin1313-bin1314 | 5.6 | 17.2—26.1 | 0.9 | 10.8 | 2013FY | First flower 13-7 |
| 22.3 | bin1313-bin1314 | 4.7 | 21.7—25.0 | 0.6 | 10.9 | 2014JP | |||
| 23.3 | bin1313-bin1314 | 7.9 | 21.1—26.3 | 0.9 | 18.1 | 2012FY | |||
| 24.3 | bin1313-bin1314 | 11.1 | 21.2—26.3 | 1.0 | 24.8 | 2014YC | |||
| 26.7 | bin1315-bin1316 | 13.9 | 23.9—28.0 | 1.3 | 28.9 | 2013JP | |||
| 32.7 | bin1320-bin1321 | 5.2 | 31.9—33.6 | 0.7 | 11.5 | 2012JP | |||
| qFT-16-2 | 16 | 66.2 | bin1359-bin1360 | 3.4 | 65.9—69.6 | 0.7 | 6.2 | 2013FY | First flower 9-3 |
| qFT-20-1 | 20 | 44.8 | bin1682-bin1683 | 10.3 | 43.8—45.4 | 1.1 | 19.1 | 2013JP | First flower 21-2 |
| qFT-20-2 | 20 | 97.3 | bin1728-bin1729 | 5.3 | 94.7—99.0 | 0.7 | 11.9 | 2014JP | Novel |
| [1] | XIA Z, WATANABE S, YAMADA T, TSUBOKURA Y, NAKASHIMA H, ZHAI H, ANAI T, SATO S, YAMAZAKI T, LU S, WU H, TABATA S, HARADA K . Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proceedings of the National Academy of Sciences of the United States of America , 2012,109(32):2155-2164. |
| [2] | KONG F J, NAN H Y, CAO D, LI Y, WU F F, WANG J L, LU S J, YUAN X H, COBER E R, ABE J, LIU B H . A new dominant gene conditions early flowering and maturity in soybean. Crop Science, 2014,54(6):2529-2535. |
| [3] | JIA H C, JIANG B J, WU C X, LU W C, HOU W S, SUN S, YAN H R, HAN T F . Maturity group classification and maturity locus geno-typing of early-maturing soybean varieties from high-latitude cold regions. PLoS ONE, 2014,9:e94139. |
| [4] | COBER E R, MOLNAR S J, CHARETTE M, VOLDENG H D . A new locus for early maturity in soybean. Crop Science, 2010,50(2):524-527. |
| [5] | ZHANG W K, WANG Y J, LUO G Z, ZHANG J S, HE C Y, WU X L, GAI J Y, CHEN S Y . QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with EST markers. Theoretical and Applied Genetics, 2004,108(6):1131-1139. |
| [6] | LIU W, KIM M Y, VAN K, LEE Y H, LI H L, LIU H L, LEE S H . QTL identification of yield-related traits and their association with flowering and maturity in soybean. Journal of Crop Science and Biotechnology, 2011,14(1):65-70. |
| [7] | COOPER R L . A delayed flowering barrier to higher soybean yields. Field Crop Research, 2003,82(1):27-35. |
| [8] | KEIM P, DIERS B W, OLSON T C, SHOEMAKER R C . RFLP mapping in soybean: Association between marker loci and variation in quantitative traits. Genetics, 1990,126(3):735-742. |
| [9] | BERNARD R L . Two major genes for time of flowering and maturity in soybeans. Crop Science, 1971,11(2):242-244. |
| [10] | BUZZELL R I . Inheritance of a soybean flowering response to fluorescent-daylength conditions. Canadian Journal of Genetics and Cytology, 1971,13(4):703-707. |
| [11] | BUZZELL R I, VOLDENG H D . Inheritance of insensitivity to long daylength. Soybean Genetics Newsletter, 1980,7(1):26-29. |
| [12] | MCBLAIN B A, BERNARD R L . A new gene affecting the time of flowering and maturity in soybeans. Journal of Heredity, 1987,78(3):160-162. |
| [13] | BONATO E R, VELLO N A . E6, a dominant gene conditioning early flowering and maturity in soybeans. Genetics and Molecular Biology, 1999,22(2):229-232. |
| [14] | COBER E R, VOLDENG H D . A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T. Crop Science, 2001,41(3):698-701. |
| [15] | SAMANFAR B, MOLNAR S J, CHARETTE M, SCHOENROCK A, DEHNE F, GOLSHANI A, BELZILE F, COBER E R . Mapping and identification of a potential candidate gene for a novel maturity locus,E10, in soybean. Theoretical and Applied Genetics, 2017,130(2):377-390. |
| [16] | RAY J D, HINSON K, MANKONO J, MALO M F . Genetic control of a long-juvenile trait in soybean. Crop Science, 1995,35(4):1001-1006. |
| [17] | WANG F F, NAN H Y, CHEN L Y, FANG C, ZHANG H Y, SU T, LI S C, CHENG Q, DONG L D, LIU B H, KONG F J, LU S J . A new dominant locus,E11, controls early flowering time and maturity in soybean. Molecular Breeding, 2019,39(5):70. |
| [18] | WATANABE S, XIA Z, HIDESHIMA R, TSUBOKURA Y, SATO S, YAMANAKA N, TAKAHASHI R, ANAI T, TABATA S, KITAMURA K, HARADA K . A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics, 2011,188(2):395-407. |
| [19] | WATANABE S, HIDESHIMA R, XIA Z, TSUBOKURA Y, SATO S, NAKAMOTO Y, YAMANAKA N, TAKAHASHI R, ISHIMOTO M, ANAI T, TABATA S, HARADA K . Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics, 2009,182(4):1251-1262. |
| [20] | LIU B, KANAZAWA A, MATSUMURA H, TAKAHASHI R, HARADA K, ABE J . Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome A gene. Genetics, 2008,180(2):995-1007. |
| [21] | ZHAO C, TAKESHIMA R, ZHU J, XU M, SATO M, WATANABE S, KANAZAWA A, LIU B H, KONG F J, YAMADA T, ABE J . A recessive allele for delayed flowering at the soybean maturity locus E9 is a leaky allele of FT2a, a FLOWERING LOCUS T ortholog. BMC Plant Biology, 2016,16:20. |
| [22] | LU S J, ZHAO X H, HU Y L, LIU S L, NAN H Y, LI X M, FANG C, CAO D, SHI X Y, KONG L P, SU T, ZHANG F G, LI S C, WANG Z, YUAN X H, COBER E R, WELLER J L, LIU B H, HOU X L, TIAN Z X, KONG F J . Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nature Genetics, 2017,49(5):773-779. |
| [23] | DISSANAYAKA A, RODRIGUEZ T O, DI S, YAN F, GITHIRI S M, RODAS F R, ABE J, TAKAHASHI R . Quantitative trait locus mapping of soybean maturity gene E5. Breeding Science, 2016,66(3):407-415. |
| [24] | 向仕华, 王吴彬, 何庆元, 杨红燕, 刘成, 邢光南, 赵团结, 盖钧镒 . 多环境下野生大豆染色体片段代换系群体农艺性状相关QTL/片段的鉴定. 中国农业科学, 2015,48(1):10-22. |
| XIANG S H, WANG W B, HE Q Y, YANG H Y, LIU C, XING G N, ZHAO T J, GAI J Y . Identification of QTL/segments related to agronomic traits using CSSL population under multiple environments. Scientia Agricultura Sinica, 2015,48(1):10-22. (in Chinese) | |
| [25] | 宋晓宇, 毛婷婷, 王立伟, 刘丽凤, 李晓那, 孙石, 韩天富 . 不同播期条件下大豆开花期性状的全基因组关联分析. 中国油料作物学报, 2018,40(4):459. |
| SONG X Y, MAO T T, WANG L W, LIU L F, LI X N, SUN S, HAN T F . Genome–wide association analysis of soybean flowering time under different sowing dates. Chinese Journal of Oil Crop Sciences, 2018, 40(4):459.(in Chinese) | |
| [26] | 张雅娟, 曹永策, 李曙光, 常芳国, 孔杰杰, 盖钧镒, 赵团结 . 夏大豆重组自交系群体 NJRIMN开花期和株高QTL定位. 大豆科学, 2018,37(6):860-865. |
| ZHANG Y J, CAO Y C, LI S G, CHANG F G, KONG J J, GAI J Y, ZHAO T J . Mapping QTL for flowering time and plant height in a summer-sowing soybean RIL population NJRIMN. Soybean Science, 2018, 37(6):860-865. (in Chinese) | |
| [27] | ORF J H, CHASE K, JARVIK T, MANSURC L M, CREGAN P B, ADLER F R, LARK K G . Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Science, 1999,39(6):1642-1651. |
| [28] | YAMANAKA N, NINOMIYA S, HOSHI M, TSUBOKURA Y, YANO M, NAGAMURA Y, SASAKI T, HARADA K . An informative linkage map of soybean reveals QTLs for flowering time, leaflet morphology and regions of segregation distortion. DNA Research, 2001,8(2):61-72. |
| [29] | LEE S, JUN T H, MICHEL A P, MIAN M A R . SNP markers linked to QTL conditioning plant height, lodging, and maturity in soybean. Euphytica, 2015,203(3):521-532. |
| [30] | MAO T T, LI J Y, WEN Z X, WU T T, WU C X, SUN S, JIANG B J, HOU W S, LI W B, SONG Q J, WANG D C, HAN T F . Association mapping of loci controlling genetic and environmental interaction of soybean flowering time under various photo-thermal conditions. BMC Genomics, 2017,18(1):415. |
| [31] | WATANABE S, TSUKAMOTO C, OSHITA T, YAMADA T, ANAI T, KAGA A . Identification of quantitative trait loci for flowering time by a combination of restriction site-associated DNA sequencing and bulked segregant analysis in soybean. Breeding Science, 2017,67(3), 277-285. |
| [32] | KONG L P, LU S J, WANG Y P, FANG C, WANG F F, NAN H Y, SU T, LI S C, ZHANG F G, LI X M, ZHAO X H, YUAN X H, LIU B H, KONG F J . Quantitative trait locus mapping of flowering time and maturity in soybean using next-generation sequencing-based analysis. Frontiers in Plant Science, 2018,9:995. |
| [33] | KIM M Y, SHIN J H, KANG Y J, SHIM S R, LEE S H . Divergence of flowering genes in soybean. Journal of Biosciences, 2012,37(5):857-870. |
| [34] | ZHAI H, LÜ S X, WANG Y Q, CHEN X, REN H X, YANG J Y, CHENG W, ZONG C M, GU H P, QIU H M, WU H Y, ZHANG X Z, CUI T T, XIA Z J . Allelic variations at four major maturity E genes and transcriptional abundance of the E1 gene are associated with flowering time and maturity of soybean cultivars. PLoS ONE, 2014,9(5):e97636. |
| [35] | 邱丽娟, 常汝镇 . 大豆种质资源描述规范和数据标准. 北京:中国农业出版社, 2006. |
| QIU L J, CHANG R Z. Descriptors and data standard for soybean (Glycine spp). Beijing:China Agriculture Press, 2006. (in Chinese) | |
| [36] | NYQUIST W E, BAKER R J . Estimation of heritability and prediction of selection response in plant-populations. Critical Reviews in Plant Sciences, 1991,10.3:235-322. |
| [37] | HAN K, JEONG H J, YANG H B, KANG S M, KWON J Y, KIM S, CHOI D, KANG B C . An ultra-high-density bin map facilitates high-throughput QTL mapping of horticultural traits in pepper (Capsicum annuum). DNA Research, 2016,23(2):81-91. |
| [38] | HUANG X H, FENG Q, QIAN Q, ZHAO Q, WANG L, WANG A, GUAN J P, FAN D L, WENG Q J, HUANG T, DONG G J, SANG T, HAN B . High-throughput genotyping by whole-genome resequencing. Genome Research, 2009,19(6):1068-1076. |
| [39] | VAN OOIJEN J W . JoinMap® 4, Software for the calculation of genetic linkage maps in experimental populations. Wageningen,Kyazma BV, 2006. |
| [40] | YANG J, HU C C, HU H, YU R D, XIA Z, YE X Z, ZHU J . QTLNetwork: Mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics, 2008,24(5):721-723. |
| [41] | WANG S, BASTEN C, ZENG Z . Windows QTL cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh,NC, 2007. |
| [42] | GUTIERREZ-GONZALEZ J J, VUONG T D, ZHONG R, YU O, LEE J D, SHANNON G, ELLERSIECK M, NGUYEN H T, SLEPER D A . Major locus and other novel additive and epistatic loci involved in modulation of isoflavone concentration in soybean seeds. Theoretical and Applied Genetics, 2011,123(8):1375-1385. |
| [43] | ZOU G H, ZHAI G W, FENG Q, YAN S, WANG A, ZHAO Q, SHAO J F, ZHANG Z P, ZOU J Q, HAN B, TAO Y Z . Identification of QTLs for eight agronomically important traits using an ultra-high- density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. Journal of Experimental Botany, 2012,63(15):5451-5462. |
| [44] | ZHANG D, LI H, WANG J, ZHANG H, HU Z, CHU S, LV H, YU D . High-density genetic mapping identifies new major loci for tolerance to low-phosphorus stress in soybean. Frontiers in Plant Science, 2016,7:372. |
| [45] | JIANG N, SHI S, SHI H, KHANZADA H, WASSAN G M, ZHU C, PENG X, YU Q, CHEN X, HE X, FU J, HU L, XU J, OUYANG L, SUN X, ZHOU D, HE H, BIAN J . Mapping QTL for seed germinability under low temperature using a new high-density genetic map of rice. Frontiers in Plant Science, 2017,8:1223. |
| [46] | ARDISSON M, RANWEZ V, BESNARD A, BESNARD A, LEROY P, POUX G, ROUMET P, VIADER V, SANTONI S, DAVID J . Genotyping by sequencing using specific allelic capture to build a high-density genetic map of durum wheat. PLoS ONE, 2016,11(5):e0154609. |
| [47] | 张传量, 简俊涛, 冯洁, 崔紫霞, 许小宛, 孙道杰 . 基于90K芯片标记的小麦芒长QTL定位. 中国农业科学, 2018,51(1):17-25. |
| ZHANG C L, JIAN J T, FENG J, CUI Z X, XU X W, SUN D J . QTL Identification for awn length based on 90K array mapping in wheat. Scientia Agricultura Sinica, 2018,51(1):17-25. (in Chinese) | |
| [48] | WANG W B, LIU M F, WANG Y F, LI X L, CHENG S X, SHU L P, YU Z P, KONG J J, ZHAO T J, GAI J Y . Characterizing two inter-specific bin maps for the exploration of the QTLs/genes that confer three soybean evolutionary traits. Frontiers in Plant Science, 2016,7:1248. |
| [49] | CHENG Y B, MA Q B, REN H L, XIA Q J, SONG E L, TAN Z Y, LI S X, ZHANG G Y, NIAN H . Fine mapping of a Phytophthora- resistance gene RpsWY in soybean(Glycine max L.) by high- throughput genome-wide sequencing. Theoretical and Applied Genetics, 2017,130(5):1041-1051. |
| [50] | WANG L, CHENG Y, MA Q, MU Y, HUANG Z, XIA Q, ZHANG G, NIAN H . QTL fine-mapping of soybean (Glycine max L.) leaf type associated traits in two RILs populations. BMC Genomics, 2019,20(1):260. |
| [51] | XU Y, CROUCH J H . Marker-assisted selection in plant breeding: From publications to practice. Crop Science, 2008,48(2):391-407. |
| [52] | WANG D L, ZHU J, LI Z K L, PATERSON A H . Mapping QTLs with epistatic effects and QTL× environment interactions by mixed linear model approaches. Theoretical and Applied Genetics, 1999,99(7/8):1255-1264. |
| [53] | YANG J, ZHU J . Methods for predicting superior genotypes under multiple environments based on QTL effects. Theoretical and Applied Genetics, 2005,110(7):1268-1274. |
| [54] | CHEN X . A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science, 2004,303(5666):2022-2025. |
| [55] | ABE M, KAYA H, WATANABE-TANEDA A, SHIBUTA M, YAMAGUCHI A, SAKAMOTO T, KURATA T, AUSIN I, ARAKI T, ALONSO‐BLANCO C . FE, a phloem specific Myb related protein, promotes flowering through transcriptional activation of FLOWERING LOCUS T and FLOWERING LOCUS T INTERACTING PROTEIN 1. The Plant Journal, 2015,83(6):1059-1068. |
| [56] | HU R B, QI G, KONG Y Z, KONG D J, GAO Q, ZHOU G K . Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biology, 2010,10(1):145. |
| [57] | KIM S G, KIM S Y, PARK C M . A membrane-associated NAC transcription factor regulates salt-responsive flowering via FLOWERING LOCUS T in Arabidopsis. Planta, 2007,226(3):647-654. |
| [58] | KONG F J, LIU B H, XIA Z J, SATO S, KIM B M, WATANABE S, YAMADA T, TABATA S, KANAZAWA A, HARADA K, ABE J . Two coordinately regulated homologs of FLOWERING LOCUS T are involved in the control of photoperiodic flowering in soybean. Plant Physiology, 2010,154(3):1220-1231. |
| [1] | 彭廷燊, 陆久焱, 吴美林, 严雨欣, 刘宏周, 南文斌, 秦小健, 李明, 龚俊义, 梁永书. 多年生水稻黄糯2号和长白7号产量相关性状的QTL分析[J]. 中国农业科学, 2026, 59(7): 1361-1379. |
| [2] | 李永娟, 张悦彤, 王艺博, 赵长江, 宋洁, 陈雪丽, 姚钦. 生物炭施用对大豆轮连作系统土壤固氮微生物nifH基因丰度及群落组成的影响[J]. 中国农业科学, 2026, 59(6): 1272-1285. |
| [3] | 刘方东, 孙磊, 王吴彬, 赵晋铭, 盖钧镒. 我国大豆种植制度的变更和生态栽培区划调整的建议[J]. 中国农业科学, 2026, 59(3): 486-498. |
| [4] | 蔡廷阳, 朱玉鹏, 李瑞东, 吴宗声, 徐一帆, 宋雯雯, 徐彩龙, 吴存祥. 苗期叶损伤对黄淮海夏大豆光合特性、荚果分布及产量形成的影响[J]. 中国农业科学, 2026, 59(2): 292-304. |
| [5] | 叶美金, 陈家婷, 周界光, 尹丽, 胡欣荣, 兰雨昕, 陈斌, 苏龙兴, 刘家君, 刘天超, 李小雨, 马建. 小麦穗密度主效QTL的鉴定、验证及其遗传效应分析[J]. 中国农业科学, 2026, 59(1): 17-28. |
| [6] | 吴琼, 谢香庭, 王磊, 牟勇, 李进伟. 转基因大豆DBN8205转化体特异性定量PCR方法的研发和验证[J]. 中国农业科学, 2026, 59(1): 29-40. |
| [7] | 陈冰嬬, 唐玉劼, 张丽霞, 周宇飞, 于淼, 石贵山, 王新鼎, 李扬, 高士杰, 陆晓春, 王鼐, 刁现民. 中国粒用杂交高粱的绿色革命[J]. 中国农业科学, 2025, 58(8): 1494-1507. |
| [8] | 严孙辉, 罗程, 陈银基, 庄昕波. 细菌纤维素协同pH偏移处理对大豆分离蛋白凝胶特性与微观结构的影响[J]. 中国农业科学, 2025, 58(6): 1210-1222. |
| [9] | 刘路平, 胡雪洁, 祁金, 陈强, 刘智, 赵田湉, 史晓蕾, 刘兵强, 孟庆民, 张孟臣, 韩天富, 杨春燕. 大豆生育期基因E1和E2的启动子克隆及其表达模式分析[J]. 中国农业科学, 2025, 58(5): 840-850. |
| [10] | 杨永庆, 胡朋举, 宋亚辉, 金欣欣, 苏俏, 王瑾. 超高油花生种质SW9721-3品质性状的QTL定位[J]. 中国农业科学, 2025, 58(4): 635-646. |
| [11] | 郑煜, 陈颐, 遆晋松, 史龙飞, 许校博, 李昱霖, 郭瑞. 烟草不同轮作模式碳足迹及经济效益评价[J]. 中国农业科学, 2025, 58(4): 733-747. |
| [12] | 李璐, 谢庄, 谢可盈, 张瀚, 赵卓文, 向奥妮, 李巧龙, 凌英华, 何光华, 赵芳明. 水稻CSSL-Z492单、双片段代换系构建及粒型QTL的遗传解析[J]. 中国农业科学, 2025, 58(3): 401-415. |
| [13] | 张琦, 薛芙珍, 杨秀洁, 姜苏洋, 黄雪娟, 马佳怡, 张哲文, 徐杰飞. 大豆烟酰胺酶GmNIC1在盐碱胁迫下的功能研究[J]. 中国农业科学, 2025, 58(24): 5128-5142. |
| [14] | 马鹤逍, 葛国龙, 张向前, 路战远, 王满秀, 戎美仁, 师晶晶, 张德健, 孙雪萍. 不同作物轮作系统对土壤易氧化有机碳和碳库活度差异性的影响[J]. 中国农业科学, 2025, 58(24): 5201-5215. |
| [15] | 高春华, 赵海军, 赵逢涛, 孔玮琳, 巨飞燕, 李宗新, 石德杨, 刘苹. 生长调节剂对玉米大豆带状间作下夏玉米茎秆特性与产量的影响[J]. 中国农业科学, 2025, 58(23): 4920-4935. |
|
||