






中国农业科学 ›› 2019, Vol. 52 ›› Issue (8): 1308-1323.doi: 10.3864/j.issn.0578-1752.2019.08.002
所属专题: 作物雄性不育
薛亚东1,杨露1,杨慧丽1,李冰1,林亚楠1,张怀胜1,郭战勇1,汤继华1,2( )
)
收稿日期:2018-12-10
接受日期:2019-02-14
出版日期:2019-04-16
发布日期:2019-04-26
通讯作者:
汤继华
作者简介:薛亚东,E-mail:基金资助:
XUE YaDong1,YANG Lu1,YANG HuiLi1,LI Bing1,LIN YaNan1,ZHANG HuaiSheng1,GUO ZhanYong1,TANG JiHua1,2()
Received:2018-12-10
Accepted:2019-02-14
Online:2019-04-16
Published:2019-04-26
Contact:
JiHua TANG
摘要:
【目的】通过分析玉米C型胞质雄性不育“三系”材料花药不同发育时期的转录组数据,以期阐明玉米C型胞质的不育和恢复机制,并解析不育基因与恢复基因之间相互作用的调控网络,为玉米C型细胞质雄性不育在不育化制种中的利用提供理论依据。【方法】以中国玉米生产上的骨干自交系豫自87-1为背景的C型胞质不育系、保持系、恢复系为材料,通过对3种材料减数分裂的前期Ⅰ、中期Ⅰ及末期Ⅱ(四分体)时期的花药进行转录组测序并利用hisat2、ballgown及DESeq2等工具进行生物信息学分析,寻找三系花药不同时期、相同时期不同材料间以及发育时序中差异表达的基因,预测C型胞质不育机制与育性恢复的调控网络;同时通过实时定量PCR对测序分析结果进行验证;通过酶活测定验证推测的C型胞质不育及恢复假说。【结果】所有材料的转录组测序共产生156.59 Gb的序列数据,比对并组装共得到53 035个基因;在恢复系与不育系、保持系与不育系以及“三系”花药不同时期之间共筛选出非重复差异基因5 676个,其中发育阶段差异基因4 705个,同时期材料间差异基因2 693个,发育时序差异基因135个。GO分子功能分析显示ATP和DNA结合相关的基因和锌离子结合基因得到高度富集;细胞组分中膜基本组分、核内及质膜内的基因得到富集;以DNA为模板的转录、转录调控、氧化还原及初级代谢等生物学过程中的基因得到富集。KEGG分析表明,差异基因主要富集于氧化磷酸化、碳代谢及糖酵解等能量代谢相关的途径中。不育系相对保持系而言,多个与氧化磷酸化相关的基因下调表达,而恢复系中不但相应基因的表达水平得到恢复,而且同时协调调节了同一能量代谢途径中的其他基因,定量分析显示差异基因的表达差异及趋势与转录组测序结果基本一致。ATP酶活结果表明不育系相比保持系,ATP酶活显著降低,恢复系中由于恢复基因的作用其活性得到大幅恢复。【结论】玉米C型胞质不育基因引起基因表达变化可能发生在减数分裂中期Ⅰ之后,末期Ⅱ之前;玉米C型胞质不育的形成可能是由于不育基因引起的能量亏损所致,而恢复基因则通过能量补偿促使育性得以恢复。
薛亚东,杨露,杨慧丽,李冰,林亚楠,张怀胜,郭战勇,汤继华. 玉米C型细胞质雄性不育花药不同发育时期的转录组分析[J]. 中国农业科学, 2019, 52(8): 1308-1323.
XUE YaDong,YANG Lu,YANG HuiLi,LI Bing,LIN YaNan,ZHANG HuaiSheng,GUO ZhanYong,TANG JiHua. Comparative Transcriptome Analysis Among the Three Line of Cytoplasmic Male Sterility in Maize[J]. Scientia Agricultura Sinica, 2019, 52(8): 1308-1323.
表1
差异基因表达分析所用引物"
| 引物名称 Primer name | 引物序列 Primer sequence (5′-3′) | 作用 Function |
|---|---|---|
| Zm00001d043834-F | GAGCAAGCTACAGAGCAGCA | Zm00001d043834表达分析 |
| Zm00001d043834-R | GCACCACCAAAGAGACCAAT | For the expression of Zm00001d043834 |
| Zm00001d009222-F | GAGATCCAGAGCGCCATTT | Zm00001d009222表达分析 |
| Zm00001d009222-R | GAGCCCAGGAAGAGGAAGAT | For the expression of Zm00001d009222 |
| Zm00001d007966-F | GTGCATCACCAAGCTCTTCC | Zm00001d007966表达分析 |
| Zm00001d007966-R | GTGCCACCTCCAATCATCTT | For the expression of Zm00001d007966 |
| Zm00001d009727-F | TTGTCTGCACGAGGAATCAG | Zm00001d009727表达表达 |
| Zm00001d009727-R | ACCAGACGACATCGTGTTCA | For the expression of Zm00001d009727 |
表2
测序数据统计分析"
| 样品 Sample | 总序列 Total read pairs | 总碱基数 Total bases (Billion) | GC含量 GC percentage (%) | Q30比例 Q30 percentage (%) | 比对序列比例 Total mapping reads percentage (%) |
|---|---|---|---|---|---|
| CrP11 | 18374016 | 5.51 | 54.5 | 94.3 | 90.0 |
| CrP12 | 16560680 | 4.96 | 54.5 | 93.7 | 91.1 |
| CrP13 | 27650293 | 6.91 | 52.5 | 100.0 | 91.3 |
| CrM11 | 17989607 | 5.39 | 54.5 | 93.3 | 90.6 |
| CrM12 | 16908690 | 5.07 | 53.5 | 95.0 | 90.7 |
| CrM13 | 26782609 | 6.69 | 52.0 | 100.0 | 91.5 |
| CrT21 | 18705720 | 5.61 | 54.5 | 93.7 | 90.2 |
| CrT22 | 17698741 | 5.31 | 54.5 | 95.0 | 90.1 |
| CrT23 | 13798055 | 2.48 | 55.0 | 80.5 | 83.6 |
| CRP11 | 22127378 | 6.63 | 54.0 | 100.0 | 90.6 |
| CRP12 | 24978644 | 7.49 | 54.0 | 100.0 | 90.8 |
| CRP13 | 27365104 | 6.84 | 52.0 | 100.0 | 91.7 |
| CRM11 | 28575880 | 8.57 | 54.0 | 100.0 | 90.4 |
| CRM12 | 18086263 | 5.42 | 55.0 | 97.6 | 89.8 |
| CRM13 | 29768530 | 7.44 | 52.5 | 100.0 | 91.3 |
| CRT21 | 27996156 | 8.39 | 55.5 | 93.3 | 90.3 |
| CRT22 | 20498349 | 6.14 | 55.5 | 94.7 | 90.1 |
| CRT23 | 12902671 | 2.32 | 55.0 | 78.9 | 83.1 |
| NrP11 | 17959776 | 5.38 | 54.0 | 94.0 | 91.0 |
| NrP12 | 18272442 | 5.48 | 55.0 | 94.7 | 90.4 |
| NrP13 | 28541683 | 7.13 | 52.0 | 100.0 | 90.8 |
| NrM11 | 19324515 | 5.79 | 55.5 | 94.0 | 89.6 |
| NrM12 | 18245189 | 5.47 | 55.5 | 94.0 | 89.8 |
| NrM13 | 29194399 | 7.29 | 52.5 | 100.0 | 92.1 |
| NrT21 | 21652022 | 6.49 | 57.0 | 100.0 | 89.4 |
| NrT22 | 20912786 | 6.27 | 56.0 | 100.0 | 90.0 |
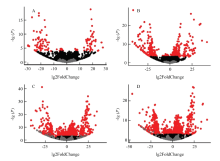
图1
差异基因火山图 A:不育系中期Ⅰ与前期Ⅰ比较;B:恢复系中期Ⅰ与前期Ⅰ比较;C:四分体时期恢复系与不育系比较;D:四分体时期保持系与不育系比较"
表3
差异基因统计分析"
| 差异比较 Comparison | 表达趋势 Up-/Down regulated | 差异基因数 DE gene numbers | 差异比较 Comparison | 表达趋势 Up-/Down regulated | 差异基因数 DE gene numbers |
|---|---|---|---|---|---|
| CrM1/CrP1 | 上调Up | 48 | CRP1/CrP1 | 上调Up | 57 |
| 下调Down | 73 | 下调Down | 160 | ||
| CrT2/CrM1 | 上调Up | 85 | CRM1/CrM1 | 上调Up | 273 |
| 下调Down | 329 | 下调Down | 380 | ||
| CrT2/CrP1 | 上调Up | 230 | CRT2/CrT2 | 上调Up | 975 |
| 下调Down | 1088 | 下调Down | 454 | ||
| CRM1/CRP1 | 上调Up | 233 | NrP1/CrP1 | 上调Up | 50 |
| 下调Down | 192 | 下调Down | 53 | ||
| CRT2/CRM1 | 上调Up | 142 | NrM1/CrM1 | 上调Up | 99 |
| 下调Down | 188 | 下调Down | 64 | ||
| CRT2/CRP1 | 上调Up | 732 | NrT2/CrT2 | 上调Up | 723 |
| 下调Down | 775 | 下调Down | 409 | ||
| NrM1/NrP1 | 上调Up | 52 | 发育时序 Time Series | NA/无 | 135 |
| 下调Down | 54 | ||||
| NrT2/NrM1 | 上调Up | 356 | NrT2/NrP1 | 上调Up | 786 |
| 下调Down | 560 | 下调Down | 1078 |
图2
差异表达GO分类 转运:Transport;翻译:Translation;DNA模板的转录:Transcription, DNA-templated;系统发育:System development;信号发射:Signaling;信号转导:Signal transduction;水缺乏响应:Response to water deprivation;胁迫响应:Response to stress;刺激物响应:Response to stimulus;盐胁迫响应:Response to salt stress;冷响应:Response to cold;化学物响应:Response to chemical;镉离子响应:Response to cadmium ion;脱落酸响应:Response to abscisic acid;转录调节:Regulation of transcription;细胞过程调节:Regulation of cellular process;生物学过程调节:Regulation of biological process;嘌呤核苷酸代谢过程:Purine ribonucleotide catabolic process;嘌呤三磷酸核苷代谢过程:Purine ribonucleoside triphosphate catabolic process;蛋白水解:Proteolysis;蛋白泛素化:Protein ubiquitination;蛋白磷酸化:Protein phosphorylation;初级代谢过程:Primary metabolic process;翻译后蛋白修饰:Post-translational protein modification;胚胎后动物器官发育:Post-embryonic animal organ development;DNA模板的转录正调控:Positive regulation of transcription, DNA-templated;磷酸化:Phosphorylation;氧化还原过程:Oxidation-reduction process;多细胞机体过程:Multicellular organismal process;多细胞生物发育:Multicellular organism development;多生物体过程:Multi-organism process;定位:Localization;基因表达:Gene expression;表皮细胞分化:Epidermal cell differentiation;发育过程:Developmental process;染色体组建:Chromosome organization;细胞过程:Cellular process;细胞氮复合物代谢过程:Cellular nitrogen compound metabolic process;细胞代谢过程:Cellular metabolic process;细胞大分子代谢过程:Cellular macromolecule metabolic process;细胞大分子生物合成过程:Cellular macromolecule biosynthetic process;细胞酮基代谢过程:Cellular ketone metabolic process;细胞组分组建:Cellular component organization;细胞生物合成过程:Cellular biosynthetic process;生物合成过程:Biosynthetic process;生物学调控:Biological regulation;动物器官发育:Animal organ development;解剖结构的形态建成:Anatomical structure morphogenesis;形态建成中解剖结构的形成:Anatomical structure formation involved in morphogenesis;液泡:Vacuole;液泡膜:Vacuolar membrane;蛋白复合体:Protein complex;胞间连丝:Plasmodesma;质膜:Plasma membrane;植物类型细胞壁:Plant-type cell wall;核:Nucleus;核仁:Nucleolus;线粒体:Mitochondrion;膜:Membrane;膜基本组分:Integral component of membrane;高尔基体装置:Golgi apparatus;细胞外区域:Extracellular region;内质网:Endoplasmic reticulum;细胞溶质:Cytosol;细胞质:Cytoplasm;叶绿体基质:Chloroplast stroma;叶绿体:Chloroplast;细胞壁:Cell wall;质外体:Apoplast;锌离子结合:Zinc ion binding;特定序列的DNA结合:Sequence-specific DNA binding;RNA结合:RNA binding;蛋白丝氨酸/苏氨酸激酶活性:Protein serine/threonine kinase activity;蛋白二聚化活性:Protein dimerization activity;核苷酸结合:Nucleotide binding;核酸结合:Nucleic acid binding;金属离子结合:Metal ion binding;DNA结合的转录因子活性:DNA binding transcription factor activity;DNA结合:DNA binding;结合:Binding;ATP结合:ATP binding。BP:生物过程 Biological processes;CC:细胞组分 Cell components;MF:分子功能 Molecular function"
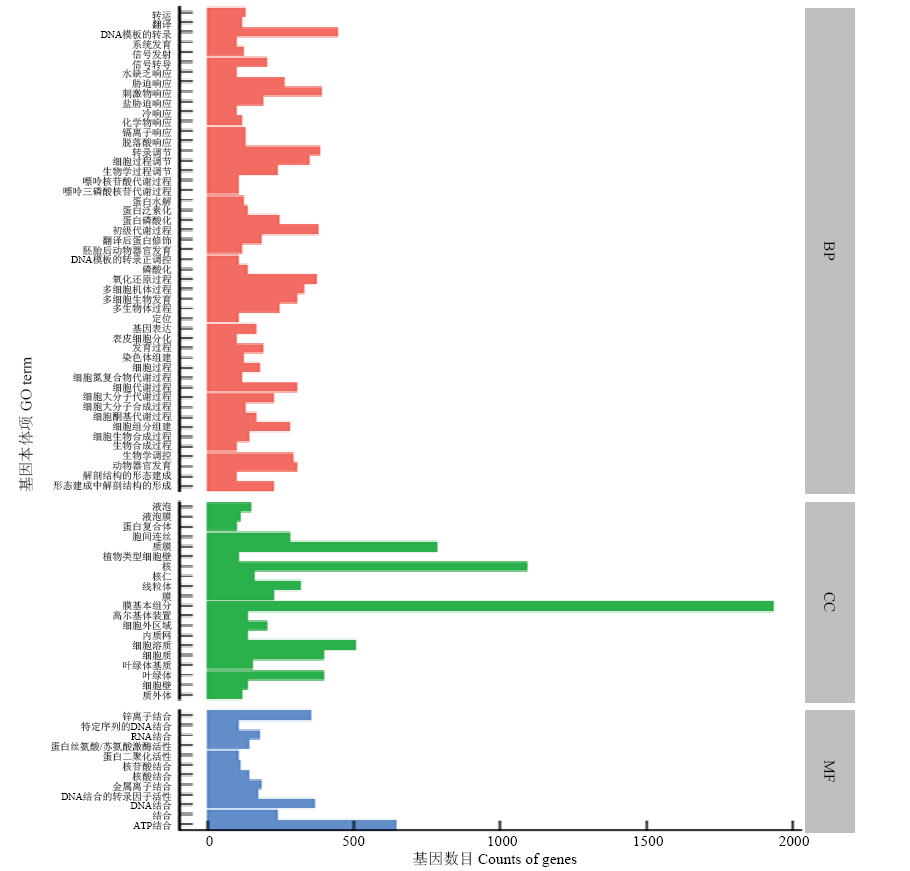
表4
差异表达基因数量最多的15个代谢通路"
| 代谢通路 Pathway ID | 通路名称 Name of pathways | 通路出现次数 Occurrences of pathways | 基因数目 Counts of genes |
|---|---|---|---|
| zma01200 | 碳代谢 Carbon metabolism | 1 | 28 |
| zma00010 | 糖酵解 Glycolysis/Gluconeogenesis | 1 | 20 |
| zma03030 | DNA复制 DNA replication | 1 | 19 |
| zma00620 | 丙酮酸盐代谢 Pyruvate metabolism | 2 | 16 |
| zma03440 | 同源重组 Homologous recombination | 1 | 15 |
| zma00071 | 脂肪酸降解 Fatty acid degradation | 5 | 14 |
| zma00130 | 辅酶Q及其他萜-醌合成 Ubiquinone and other terpenoid-quinone biosynthesis | 6 | 13 |
| zma00040 | 戊糖与葡萄糖醛酸相互转换 Pentose and glucuronate interconversions | 1 | 9 |
| zma00073 | 角质、软木脂及蜡质合成 Cutin, suberine and wax biosynthesis | 12 | 8 |
| zma00940 | 苯丙烷合成 Phenylpropanoid biosynthesis | 5 | 6 |
| zma00290 | 缬氨酸、亮氨酸和异亮氨酸合成 Valine, leucine and isoleucine biosynthesis | 2 | 5 |
| zma02010 | ABC运输 ABC transporters | 1 | 5 |
| zma00500 | 淀粉和蔗糖代谢 Starch and sucrose metabolism | 1 | 4 |
| zma00770 | 泛酸酯和辅酶A合成 Pantothenate and CoA biosynthesis | 1 | 4 |
| zma04016 | 植物MAPK信号途径 MAPK signaling pathway-plant | 1 | 4 |
表5
线粒体氧化磷酸化途径相关基因的表达差异"
| 基因编号 ID number of gene | 中期Ⅰ恢复系比不育系 CR/Cr-m1 | 四分体恢复系比不育系 CR/Cr-t2 | 四分体保持系比不育系 Nr/Cr-t2 | 不育系四分体比前期Ⅰ Cr-t2/p1 | 保持系四分体比中期Ⅰ Nr-t2/m1 | 保持系四分体比前期Ⅰ Nr-t2/p1 | 恢复系四分体比前期Ⅰ CR-t2/p1 | 染色体 Chr. | 基因功能描述 Description |
|---|---|---|---|---|---|---|---|---|---|
| Zm00001d033552 | 2.94±0.83 | 3.60±0.83 | 3.78±0.93 | 4.09±0.93 | 5.95±0.93 | 5.15±0.83 | 1 | 腺苷三磷酸酶4, 质膜类型 ATPase 4 plasma membrane-type | |
| Zm00001d043834 | 1.33±0.40 | 1.44±0.40 | 1.42±0.40 | 1.07±0.35 | 3 | 类线粒体腺苷三磷酸合成酶贝塔亚基 ATP synthase subunit beta, mitochondrial-like | |||
| Zm00001d045122 | 4.60±1.08 | 4.04±1.19 | 9 | 腺苷三磷酸酶8, 质膜类型 ATPase 8 plasma membrane-type | |||||
| Zm00001d041214 | 1.74±0.54 | 3 | 腺苷三磷酸酶4, 质膜类型 ATPase 4 plasma membrane-type | ||||||
| Zm00001d015569 | 1.64±0.41 | 5 | 液泡质子泵同系物1 Vacuolar proton pump homolog 1 | ||||||
| Zm00001d037576 | 2.22±0.6 | 2.54±0.67 | -1.85±0.60 | 6 | 焦磷酸供能的液泡质膜质子泵 Pyrophosphate-energized vacuolar membrane proton pump | ||||
| Zm00001d009222 | 3.38±1.07 | -3.61±1.06 | -7.12±1.77 | -7.90±1.77 | 8 | 焦磷酸供能的液泡质膜质子泵 Pyrophosphate-energized vacuolar membrane proton pump | |||
| Zm00001d052022 | -5.78±1.58 | 4 | 腺苷三磷酸酶8, 质膜类型 ATPase 8 plasma membrane-type | ||||||
| Zm00001d045497 | -2.04±0.59 | 9 | V型质子偶联腺苷三磷酸亚基e1 V-type proton ATPase subunit e1 | ||||||
| Zm00001d027304 | -3.2±0.90 | -2.94±1.00 | -3.17±0.90 | 1 | 腺苷三磷酸酶9, 质膜类型 ATPase 9, plasma membrane-type | ||||
| Zm00001d007966 | -1.18±0.30 | -1.17±0.30 | 2 | 琥珀酸脱氢酶4 Succinate dehydrogenase4 | |||||
| Zm00001d036728 | -4.01±0.95 | 6 | 腺苷三磷酸酶2, 质膜类型 ATPase 2, plasma membrane-type | ||||||
| Zm00001d009727 | -8.63±2.86 | 8 | 线粒体细胞色素c氧化酶亚基5b-2 Cytochrome c oxidase subunit 5b-2 mitochondrial |
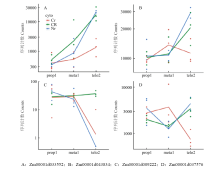
图3
发育过程中基因表达分析"
图4
氧化磷酸化途径中差异表达基因 绿底框:物种特异的基因;红底框:上调基因;天蓝底框:下调基因"
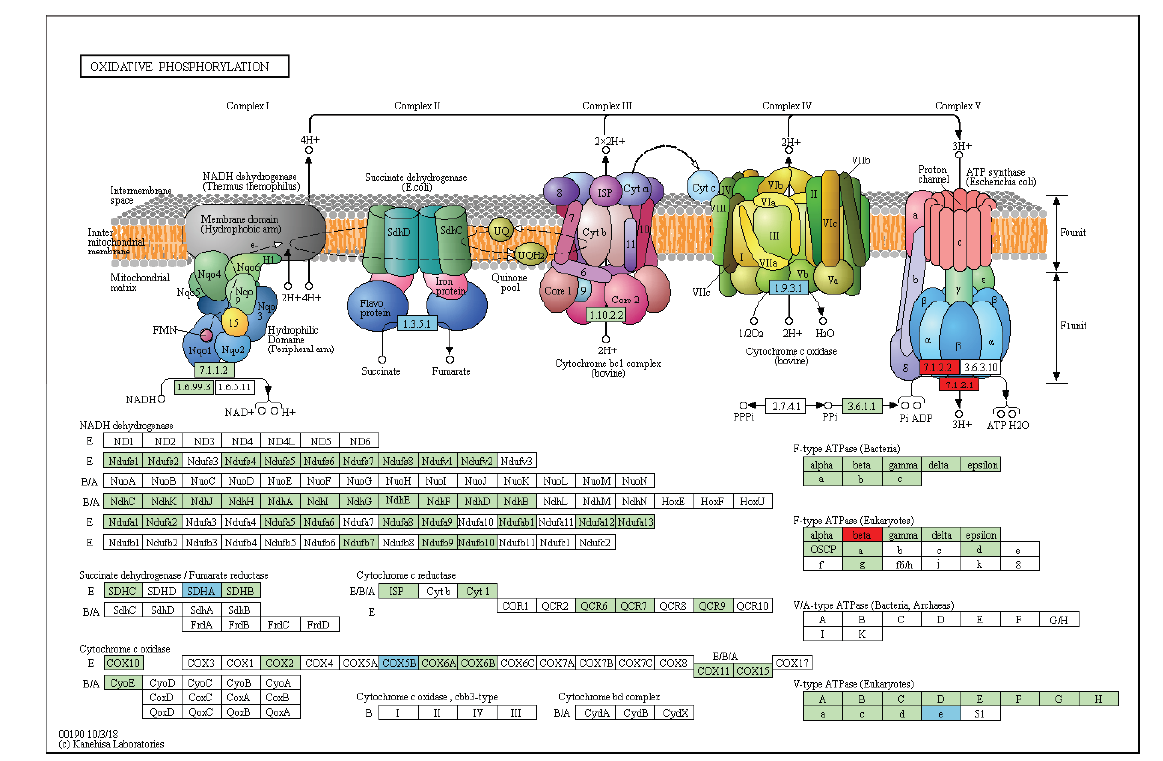
图5
不同发育时期差异基因荧光定量检测 CR:恢复系;Cr:不育系;Nr:保持系,M1:中期1;T2:末期2(tetrad);MN:单核期"
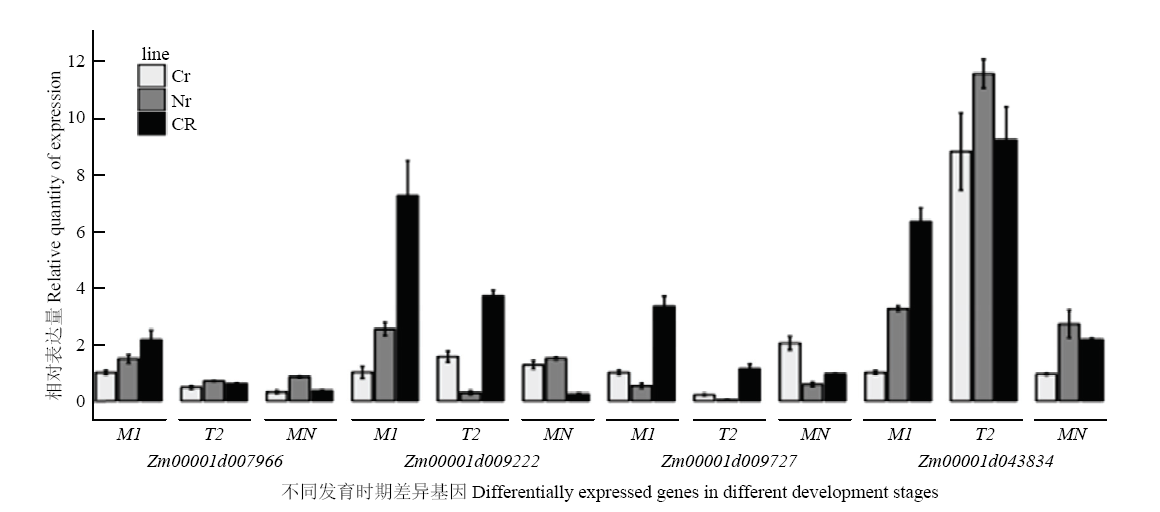
表6
细胞色素P450基因的表达"
| 时期 stage | 比较 Comparison | 表达 Expression | 差异倍数log2FLC | 基因ID Gene ID | 染色体 Chr. | 功能描述 Description |
|---|---|---|---|---|---|---|
| 末期2 Telo2 | Nr/Cr | 上调Up | 21.83 | Zm00001d024412 | 10 | NADPH-细胞色素P450还原酶 NADPH-cytochrome P450 reductase |
| 9.20 | Zm00001d042814 | 3 | 86A1型细胞色素P450 Cytochrome P450 86A1 | |||
| 7.31 | Zm00001d011932 | 8 | 类89A2型细胞色素P450 Cytochrome P450 89A2-like | |||
| 6.94 | Zm00001d013862 | 5 | 细胞色素P450大家族蛋白 Cytochrome P450 superfamily protein | |||
| 6.92 | Zm00001d029526 | 1 | 78A5型细胞色素P450 Cytochrome P450 78A5 | |||
| 下调Down | -17.05 | Zm00001d039697 | 3 | 类711A1型细胞色素P450 Cytochrome P450 711A1-like | ||
| -20.91 | Zm00001d004486 | 2 | 71D7型细胞色素P450 Cytochrome P450 71D7 | |||
| CR/Cr | 上调Up | 18.54 | Zm00001d024412 | 10 | 细胞色素P450还原酶 Cytochrome P450 reductase | |
| 8.54 | Zm00001d042814 | 3 | 86A1型细胞色素P450 Cytochrome P450 86A1 | |||
| 7.29 | Zm00001d013862 | 5 | 推断的细胞色素P450大家族蛋白 Putative cytochrome P450 superfamily protein | |||
| 7.23 | Zm00001d011932 | 8 | 类89A2型细胞色素P450 Cytochrome P450 89A2-like | |||
| 6.46 | Zm00001d012326 | 8 | 细胞色素P450大家族蛋白 Cytochrome P450 superfamily protein | |||
| 5.82 | Zm00001d029526 | 1 | 78A5型细胞色素P450 Cytochrome P450 78A5 | |||
| 下调Down | -1.22 | Zm00001d002937 | 2 | 细胞色素P450,72家族,A亚族,多肽8 Cytochrome P450 family 72 subfamily A polypeptide 8 | ||
| -20.04 | Zm00001d039697 | 3 | 类711A1型细胞色素P450 Cytochrome P450 711A1-like | |||
| -23.94 | Zm00001d004486 | 2 | 71D7型细胞色素P450 Cytochrome P450 71D7 | |||
| 中期1 Meta1 | Nr/Cr | 上调Up | - | - | - | - |
| 下调Down | - | - | - | - | ||
| CR/Cr | 上调Up | 7.46 | Zm00001d011932 | 8 | 类89A2型细胞色素P450 Cytochrome P450 89A2-like | |
| 6.79 | Zm00001d013862 | 5 | 推断的细胞色素P450大家族蛋白 Putative cytochrome P450 superfamily protein | |||
| 3.88 | Zm00001d020673 | 7 | 推断的细胞色素P450大家族蛋白 Putative cytochrome P450 superfamily protein | |||
| 下调Down | -3.58 | Zm00001d049573 | 4 | 推断的细胞色素P450大家族蛋白 Putative cytochrome P450 superfamily protein | ||
| -4.16 | Zm00001d037701 | 6 | 推断的细胞色素P450大家族蛋白 Putative cytochrome P450 superfamily protein | |||
| -16.87 | Zm00001d039697 | 3 | 类711A1型细胞色素P450 Cytochrome P450 711A1-like | |||
| 前期1 Prop1 | Nr/Cr | 上调Up | 19.96 | Zm00001d024412 | 10 | NADPH-细胞色素P450还原酶 NADPH--cytochrome P450 reductase |
| 17.59 | Zm00001d042814 | 3 | 86A1型细胞色素P450 Cytochrome P450 86A1 | |||
| 下调Down | - | - | - | - | ||
| CR/Cr | 上调Up | 17.44 | Zm00001d042814 | 3 | 86A1型细胞色素P450 Cytochrome P450 86A1 | |
| 下调Down | - | - | - | - |
图6
三系材料花药ATP酶活性测定与比较 CR:恢复系;Cr:不育系;Nr:保持系;**表示0.01水平差异显著;***表示0.001水平差异显著"
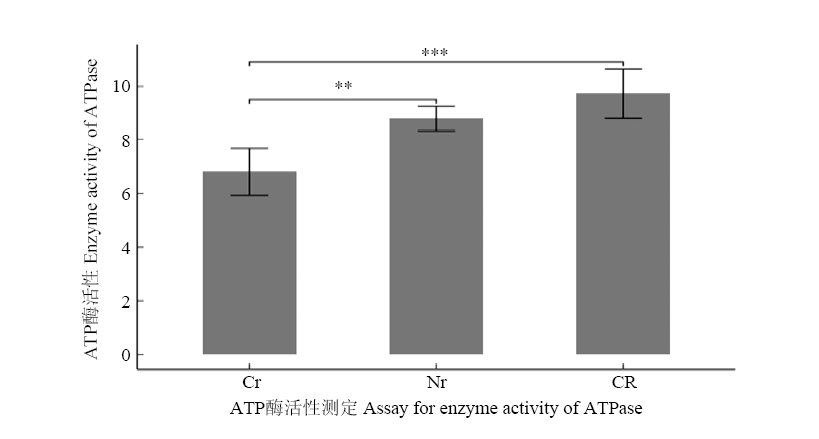
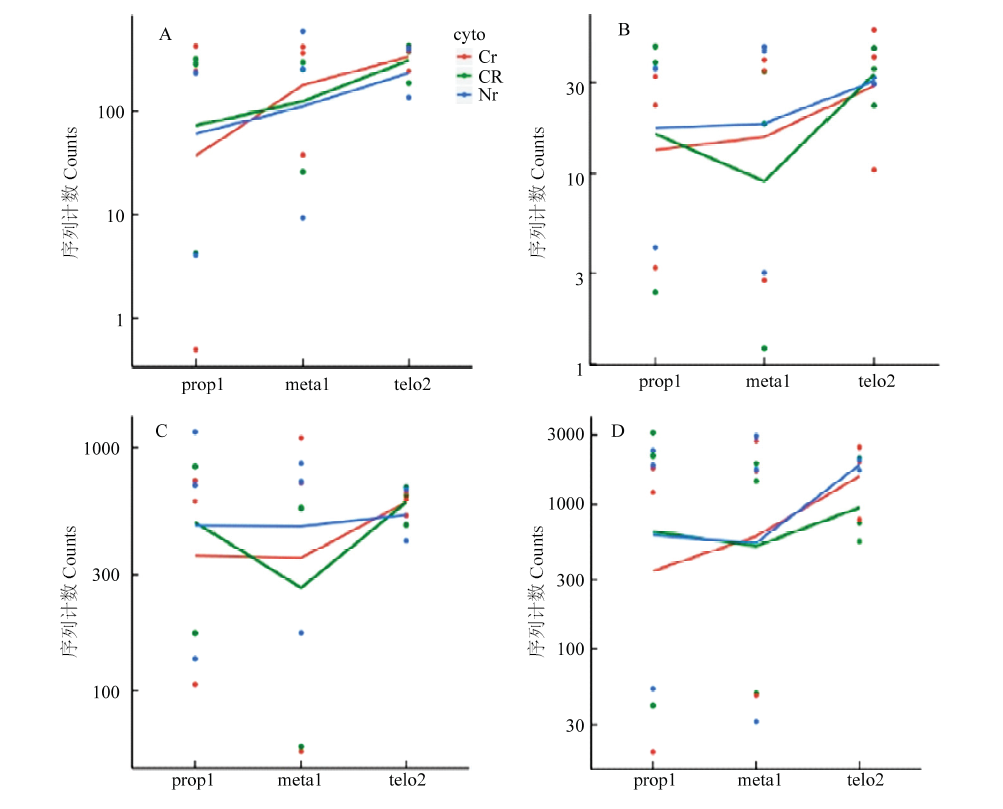
图7
线粒体基因atp6c、atp9-1、cox2-1及nad4时序表达分析"
| [1] |
DEWEY R, LEVINGS C S, TIMOTHY D H . Novel recombinations in the maize mitochondrial genome produce a unique transcriptional unit in the texas male-sterile cytoplasm. Cell, 1986,44(3):439-449.
doi: 10.1016/0092-8674(86)90465-4 |
| [2] |
PENG X, WANG K, HU C, ZHU Y, WANG T, YANG J, TONG J, LI S, ZHU Y . The mitochondrial gene orfH79 plays a critical role in impairing both male gametophyte development and root growth in CMS-Honglian rice. BMC Plant Biology, 2010,10(1):125.
doi: 10.1186/1471-2229-10-125 |
| [3] |
WANG Z, ZOU Y, LI X, ZHANG Q, CHEN L, WU H, SU D, CHEN Y, GUO J, LUO D, LONG Y, ZHONG Y, LIU Y G . Cytoplasmic male sterility of rice with Boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. The Plant Cell, 2006,18(3):676-687.
doi: 10.1105/tpc.105.038240 |
| [4] | LUO D, XU H, LIU Z, GUO J, LI H, CHEN L, FANG C, ZHANG Q, BAI M, YAO N, WU H, WU H, JI C, ZHENG H, CHEN Y, YE S, LI X, ZHAO X, LI R, LIU Y G . A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nature Genetics, 2013,45(5):573-577. |
| [5] |
SINGH M . Suppression of cytoplasmic male sterility by nuclear genes alters expression of a novel mitochondrial gene region. The Plant Cell, 1991,3(12):1349-1362.
doi: 10.1105/tpc.3.12.1349 |
| [6] | BROWN G G . Unique aspects of cytoplasmic male sterility and fertility restoration in Brassica napus. Journal of Heredity, 1999,90(3):351-356. |
| [7] | UYTTEWAAL M, ARNAL N, QUADRADO M, MARTIN-CANADELL A, VRIELYNCK N, HIARD S, GHERBI H, BENDAHMANE A, BUDAR F, MIREAU H . Characterization of Raphanus sativus pentatricopeptide repeat proteins encoded by the fertility restorer locus for Ogura cytoplasmic male sterility. The Plant Cell, 2008,20(12):3331-3345. |
| [8] | CUI X, WISE R P, SCHNABLE P S . The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science, 1996,272(5266):1334-1336. |
| [9] |
FUJII S, TORIYAMA K . Suppressed expression of retrograde- regulated male sterility restores pollen fertility in cytoplasmic male sterile rice plants. Proceedings of the National Academy of Sciences of the USA, 2009,106(23):9513-9518.
doi: 10.1073/pnas.0901860106 |
| [10] | ITABASHI E, IWATA N, FUJII S, KAZAMA T, TORIYAMA K . The fertility restorer gene,Rf2, for lead rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. The Plant Journal, 2011,65(3):359-367. |
| [11] | KITAZAKI K, ARAKAWA T, MATSUNAGA M, YUI-KURINO R, MATSUHIRA H, MIKAMI T, KUBO T . Post-translational mechanisms are associated with fertility restoration of cytoplasmic male sterility in sugar beet (Beta vulgaris). The Plant Journal, 2015,83(2):290-299. |
| [12] |
BENTOLILA S, ALFONSO A A, HANSON M R . A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. Proceedings of the National Academy of Sciences of the USA, 2002,99(16):10887-10892.
doi: 10.1073/pnas.102301599 |
| [13] | BROWN G G, FORMANOVÁ N, JIN H, WARGACHUK R, DENDY C, PATIL P, LAFOREST M, ZHANG J, CHEUNG W Y, LANDRY B S . The radish Rfo restorer gene of Ogura cytoplasmic male sterility encodes a protein with multiple pentatricopeptide repeats. The Plant Journal, 2003,35(2):262-272. |
| [14] | KOIZUKA N, IMAI R, FUJIMOTO H, HAYAKAWA T, KIMURA Y, KOHNO-MURASE J, SAKAI T, KAWASAKI S, IMAMURA J . Genetic characterization of a pentatricopeptide repeat protein gene,orf687, that restores fertility in the cytoplasmic male-sterile Kosena radish. The Plant Journal, 2003,34(4):407-415. |
| [15] | HU J, WANG K, HUANG W, LIU G, GAO Y, WANG J, HUANG Q, JI Y, QIN X, WAN L, ZHU R, LI S, YANG D, ZHU Y . The rice pentatricopeptide repeat protein Rf5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. The Plant Cell, 2012,24(1):109-122. |
| [16] | TANG H, LUO D, ZHOU D, ZHANG Q, TIAN D, ZHENG X, CHEN L, LIU Y G . The rice restorer Rf4 for wild-abortive cytoplasmic male sterility encodes a mitochondrial-localized PPR protein that functions in reduction of WA352 transcripts. Molecular Plant, 2014,7(9):1497-1500. |
| [17] |
HUANG W, YU C, HU J, WANG L, DAN Z, ZHOU W, HE C, ZENG Y, YAO G, QI J, ZHANG Z, ZHU R, CHEN X, ZHU Y . Pentatricopeptide-repeat family protein RF6 functions with hexokinase 6 to rescue rice cytoplasmic male sterility. Proceedings of the National Academy of Sciences of the USA, 2015,112(48):14984-14989.
doi: 10.1073/pnas.1511748112 |
| [18] |
CHEN L, LIU Y G . Male sterility and fertility restoration in crops. Annual Review of Plant Biology, 2014,65(1):579-606.
doi: 10.1146/annurev-arplant-050213-040119 |
| [19] | 陈伟程, 罗福和, 季良越 . 玉米C型胞质雄花不育的遗传及其在生产上的应用. 作物学报, 1979,5(4):21-28. |
| CHEN W C, LUO F H, JI L Y . Some genetic aspects of the C-type cytoplasmic male-sterility in maize and its use in breeding. Acta Agronomica Sinica, 1979,5(4):21-28. (in Chinese) | |
| [20] | 汤继华, 刘宗华, 陈伟程, 胡彦民, 季洪强, 季良越 . 玉米C型胞质不育恢复主基因SSR标记. 中国农业科学, 2001,34(6):592-596. |
| TANG J H, LIU Z H, CHEN W C, HU Y M, JI H Q, JI L Y . The SSR markers of the main restorer genes for CMS-C cytoplasmic male sterility in maize. Scientia Agricultura Sinica, 2001,34(6):592-596. (in Chinese) | |
| [21] |
DEWEY R E, TIMOTHY D H, LEVINGS C S . Chimeric mitochondrial genes expressed in the C male-sterile cytoplasm of maize. Current Genetics, 1991,20(6):475-482.
doi: 10.1007/BF00334775 |
| [22] |
ALLEN J O, FAURON C M, MINX P, ROARK L, ODDIRAJU S, LIN G N, MEYER L, SUN H, KIM K, WANG C, DU F, XU D, GIBSON M, CIFRESE J, CLIFTON S W, NEWTON K J . Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics, 2007,177(2):1173-1192.
doi: 10.1534/genetics.107.073312 |
| [23] | LEE S L, GRACEN V E, EARLE E D . The cytology of pollen abortion in C-cytoplasmic male-sterile corn anthers. American Journal of Botany, 1979,66(6):12. |
| [24] | 陈伟程, 李桂珍 . 玉米C型胞质雄性不育系花粉败育的细胞学研究. 华北农学报, 1987,2(1):1-6. |
| CHEN W C, LI G Z . A cytological study in pollen abortion in C-cytoplasmic male-sterile corn (Zea mays, L.). Acta Agriculturae Boreali-Sinica, 1987,2(1):1-6. (in Chinese) | |
| [25] | LIU Q, LAN Y, WEN C, ZHAO H, WANG J, WANG Y . Transcriptome sequencing analyses between the cytoplasmic male sterile line and its maintainer line in Welsh onion (Allium fistulosum L.). International Journal of Molecular Sciences, 2016,17(7):1058. |
| [26] | LIU C, MA N, WANG P Y, FU N, SHEN H L . Transcriptome sequencing and De Novo analysis of a cytoplasmic male sterile line and its near-isogenic restorer line in Chili pepper (Capsicum annuum L.). PLoS ONE, 2013,8(6):e65209. |
| [27] | LI C, ZHAO Z, LIU Y, LIANG B, GUAN S, LAN H, WANG J, LU Y, CAO M . Comparative transcriptome analysis of isonuclear-alloplasmic lines unmask key transcription factor genes and metabolic pathways involved in sterility of maize CMS-C. PeerJ-the Journal of Life and Environmental Science, 2017,5:e3408. |
| [28] |
MA J, SKIBBE D S, FERNANDES J, WALBOT V . Male reproductive development: gene expression profiling of maize anther and pollen ontogeny. Genome Biology, 2008,9(12):R181.
doi: 10.1186/gb-2008-9-12-r181 |
| [29] | KIM D, LANGMEAD B, SALZBERG S L . HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 2015,12(4):357-360. |
| [30] |
PERTEA M, PERTEA G M, ANTONESCU C M, CHANG T C, MENDELL J T, SALZBERG S L . StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology, 2015,33(3):290-295.
doi: 10.1038/nbt.3122 |
| [31] |
LOVE M I, HUBER W, ANDERS S . Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 2014,15(12):550.
doi: 10.1186/s13059-014-0550-8 |
| [32] |
YU G, WANG L G, HAN Y, HE Q Y . ClusterProfiler: An R package for comparing biological themes among gene clusters. A Journal of Integrative Biology, 2012,16(5):284-287.
doi: 10.1089/omi.2011.0118 |
| [33] |
LIVAK K J, SCHMITTGEN T D . Analysis of relative gene expression data using real-time quantitative PCR and the 2 -ΔΔCT method . Methods, 2001,25(4):402-408.
doi: 10.1006/meth.2001.1262 |
| [34] |
HU J, HUANG W C, HUANG Q, QIN X J, YU C C, WANG L L, LI S Q, ZHU R S, ZHU Y G . Mitochondria and cytoplasmic male sterility in plants. Mitochondrion, 2014,19(B):282-288.
doi: 10.1016/j.mito.2014.02.008 |
| [35] |
TOUZET P, MEYER E H . Cytoplasmic male sterility and mitochondrial metablolism in plants. Mitochondrion, 2014,19(B):166-171.
doi: 10.1016/j.mito.2014.04.009 |
| [36] | WERCK-REICHHART D, FEYEREISEN R . Cytochromes P450: A success story. Genome biology, 2000, 1(6): reviews3003.1-3003.9. |
| [37] |
XU J, WANG X Y, GUO W Z . The cytochrome P450 superfmily: Key players in plant development and defense. Journal of Integrative Agriculture, 2015,14(9):1673-1686.
doi: 10.1016/S2095-3119(14)60980-1 |
| [38] |
QIAN W F, ZHANG J Z . Gene dosage and gene duplicability. Genetics, 2008,179(4):2319-2324.
doi: 10.1534/genetics.108.090936 |
| [39] | SPADAFORA N, PERROTTA L, NIEUWLAND J, ALBANI D, BITONTI B M, HERBERT R J, DOONAN J H, MARCHBANK A M, SICILIANO I, GRØNLUND A L, FRANCIS D, ROGERS H J, . Gene dosage effect of WEE1 on growth and morphogenesis from Arabidopsis hpocotyl explants. Annals of Botany, 2012,110(8):1631-1639. |
| [40] | CHANG N, SUN Q Q, LI Y Q, MU Y J, HU J L, FENG Y, LIU X M, GAO H B . PDV2 has a dosage effect on chloroplast division in Arabidopsis. Plant Cell Reports, 2017,36(3):471-480. |
| [41] | KELLIHER T, WALBOT V . Emergence and patterning of the five cell types of the Zea mays anther locule. Developmental Biology, 2011,350(1):32-49. |
| [42] | FANG W, WANG Z, CUI R, LI J, LI Y . Maternal control of seed size by EOD3/CYP78A6 in Arabidopsis thaliana. The Plant Journal, 2012,70:929-939. |
| [43] | WANG J W, SCHWAB R, CZECH B, MICA E, WEIGEL D . Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. The Plant Cell, 2008,20:1231-1243. |
| [44] | SOTELO-SILVEIRA M, CUCINOTTA M, CHAUVIN A L , CHAVEZ MONTES R A, COLOMBO L, MARSCH-MARTINEZ N, DE FOLTER S. Cytochrome P450 CYP78A9 is involved in Arabidopsis reproductive development. Plant Physiology, 2013,162:779-799. |
| [45] |
LI H, PINOT F, SAUVEPLANE V, WERCK-REICHHART D, DIEHL P, SCHREIBER L, FRANKE R, ZHANG P, CHEN L, GAO Y W, LIANG W Q, ZHANG D B . Cytochrome P450 family member CYP704B2 catalyzes the omega-hydroxylation of fatty acids and is required for anther cutin biosynthesis and pollen exine formation in rice. The Plant Cell, 2010,22:173-190.
doi: 10.1105/tpc.109.070326 |
| [46] |
DJUKANOVIC V, SMITH J, LOWE K, YANG M Z, GAO H R, JONES S, MICHOLSON M G, WEST A, LAPE J, BIDNEY D, FALCO S C, JANTZ D, LYZNIK L A . Male-sterile maize plants produced by targeted mutagenesis of the cytochrome P450-like gene (MS26) using a re-designed I-CreI homeing endonuclease. The Plant Journal, 2013,76:888-899.
doi: 10.1111/tpj.12335 |
| [1] | 赵政鑫,王晓云,田雅洁,王锐,彭青,蔡焕杰. 未来气候条件下秸秆还田和氮肥种类对夏玉米产量及土壤氨挥发的影响[J]. 中国农业科学, 2023, 56(1): 104-117. |
| [2] | 柴海燕,贾娇,白雪,孟玲敏,张伟,金嵘,吴宏斌,苏前富. 吉林省玉米穗腐病致病镰孢菌的鉴定与部分菌株对杀菌剂的敏感性[J]. 中国农业科学, 2023, 56(1): 64-78. |
| [3] | 李周帅,董远,李婷,冯志前,段迎新,杨明羡,徐淑兔,张兴华,薛吉全. 基于杂交种群体的玉米产量及其配合力的全基因组关联分析[J]. 中国农业科学, 2022, 55(9): 1695-1709. |
| [4] | 熊伟仡,徐开未,刘明鹏,肖华,裴丽珍,彭丹丹,陈远学. 不同氮用量对四川春玉米光合特性、氮利用效率及产量的影响[J]. 中国农业科学, 2022, 55(9): 1735-1748. |
| [5] | 李易玲,彭西红,陈平,杜青,任俊波,杨雪丽,雷鹿,雍太文,杨文钰. 减量施氮对套作玉米大豆叶片持绿、光合特性和系统产量的影响[J]. 中国农业科学, 2022, 55(9): 1749-1762. |
| [6] | 马小艳,杨瑜,黄冬琳,王朝辉,高亚军,李永刚,吕辉. 小麦化肥减施与不同轮作方式的周年养分平衡及经济效益分析[J]. 中国农业科学, 2022, 55(8): 1589-1603. |
| [7] | 李前,秦裕波,尹彩侠,孔丽丽,王蒙,侯云鹏,孙博,赵胤凯,徐晨,刘志全. 滴灌施肥模式对玉米产量、养分吸收及经济效益的影响[J]. 中国农业科学, 2022, 55(8): 1604-1616. |
| [8] | 张家桦,杨恒山,张玉芹,李从锋,张瑞富,邰继承,周阳晨. 不同滴灌模式对东北春播玉米籽粒淀粉积累及淀粉相关酶活性的影响[J]. 中国农业科学, 2022, 55(7): 1332-1345. |
| [9] | 谭先明,张佳伟,王仲林,谌俊旭,杨峰,杨文钰. 基于PLS的不同水氮条件下带状套作玉米产量预测[J]. 中国农业科学, 2022, 55(6): 1127-1138. |
| [10] | 冯宣军, 潘立腾, 熊浩, 汪青军, 李静威, 张雪梅, 胡尔良, 林海建, 郑洪建, 卢艳丽. 南方地区120份甜、糯玉米自交系重要目标性状和育种潜力分析[J]. 中国农业科学, 2022, 55(5): 856-873. |
| [11] | 刘苗,刘朋召,师祖姣,王小利,王瑞,李军. 氮磷配施下夏玉米临界氮浓度稀释曲线的构建与氮营养诊断[J]. 中国农业科学, 2022, 55(5): 932-947. |
| [12] | 乔远,杨欢,雒金麟,汪思娴,梁蓝月,陈新平,张务帅. 西北地区玉米生产投入及生态环境风险评价[J]. 中国农业科学, 2022, 55(5): 962-976. |
| [13] | 黄兆福, 李璐璐, 侯梁宇, 高尚, 明博, 谢瑞芝, 侯鹏, 王克如, 薛军, 李少昆. 不同种植区玉米生理成熟后田间站秆脱水的积温需求[J]. 中国农业科学, 2022, 55(4): 680-691. |
| [14] | 石习, 宁丽华, 葛敏, 邬奇, 赵涵. 玉米氮状况相关生物标记物的筛选和应用[J]. 中国农业科学, 2022, 55(3): 438-450. |
| [15] | 张建军, 党翼, 赵刚, 王磊, 樊廷录, 李尚中. 覆膜时期和施氮量对陇东旱塬玉米产量和水氮利用效率的影响[J]. 中国农业科学, 2022, 55(3): 479-490. |
|
||