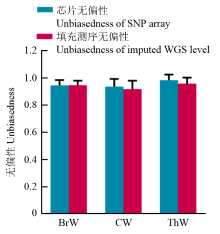
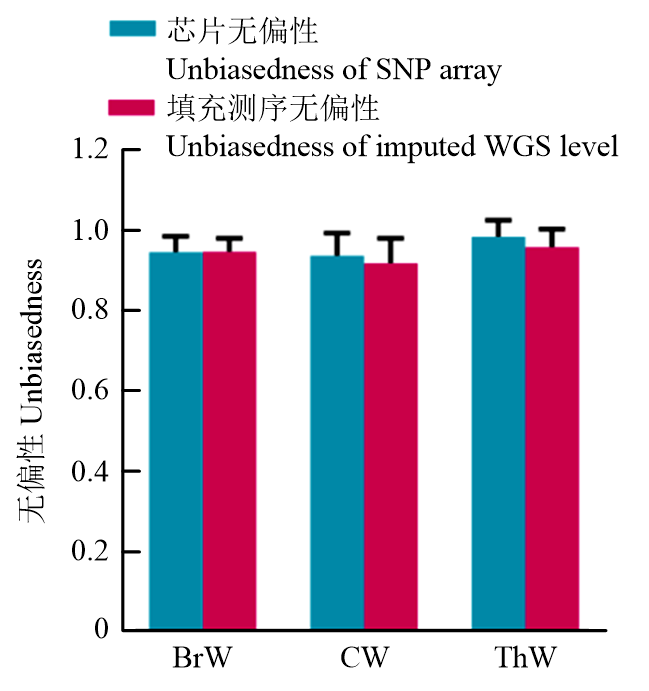
| [1] |
朱墨, 郑麦青, 崔焕先, 赵桂苹, 刘杨. 基于GBLUP和Bayes B方法对肉鸡屠宰性状基因组预测准确性的比较. 中国农业科学, 2021, 54(23): 5125-5131.
doi: 10.3864/j.issn.0578-1752.2021.23.016
|
|
ZHU M, ZHENG M Q, CUI H X, ZHAO G P, LIU Y. Comparison of genomic prediction accuracy for meat type chicken carcass traits based on GBLUP and BayesB method. Scientia Agricultura Sinica, 2021, 54(23): 5125-5131. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2021.23.016
|
| [2] |
LIU J, ZHOU J, LI J, BAO H. Identification of candidate genes associated with slaughter traits in F2 chicken population using genome-wide association study. Animal Genetics, 2021, 52(4): 532-535.
doi: 10.1111/age.v52.4
|
| [3] |
VANRADEN P M, O'CONNELL J R, WIGGANS G R, WEIGEL K A. Genomic evaluations with many more genotypes. Genetics, Selection, Evolution, 2011, 43(1): 10.
doi: 10.1186/1297-9686-43-10
|
| [4] |
BENEDICT M N, MUNDY M B, HENRY C S, CHIA N, PRICE N D. Likelihood-based gene annotations for gap filling and quality assessment in genome-scale metabolic models. PLoS Computational Biology, 2014, 10(10): e1003882.
doi: 10.1371/journal.pcbi.1003882
|
| [5] |
MEUWISSEN T, GODDARD M. Accurate prediction of genetic values for complex traits by whole-genome resequencing. Genetics, 2010, 185(2): 623-631.
doi: 10.1534/genetics.110.116590
pmid: 20308278
|
| [6] |
IHESHIULOR O O M, WOOLLIAMS J A, YU X J, WELLMANN R, MEUWISSEN T H E. Within- and across-breed genomic prediction using whole-genome sequence and single nucleotide polymorphism panels. Genetics, Selection, Evolution, 2016, 48: 15.
doi: 10.1186/s12711-016-0193-1
pmid: 26895843
|
| [7] |
MISZTAL I, LOURENCO D, LEGARRA A. Current status of genomic evaluation. Journal of Animal Science, 2020, 98(4): skaa101.
doi: 10.1093/jas/skaa101
|
| [8] |
HEIDARITABAR M, CALUS M P L, MEGENS H J, VEREIJKEN A, GROENEN M A M, BASTIAANSEN J W M. Accuracy of genomic prediction using imputed whole-genome sequence data in white layers. Journal of Animal Breeding and Genetics, 2016, 133(3): 167-179.
doi: 10.1111/jbg.12199
pmid: 26776363
|
| [9] |
HAYES B J, DAETWYLER H D. 1000 bull genomes project to map simple and complex genetic traits in cattle: Applications and outcomes. Annual Review of Animal Biosciences, 2019, 7: 89-102.
doi: 10.1146/annurev-animal-020518-115024
pmid: 30508490
|
| [10] |
KHATKAR M S, MOSER G, HAYES B J, RAADSMA H W. Strategies and utility of imputed SNP genotypes for genomic analysis in dairy cattle. BMC Genomics, 2012, 13: 538.
doi: 10.1186/1471-2164-13-538
pmid: 23043356
|
| [11] |
BHUIYAN M S A, KIM Y K, KIM H J, LEE D H, LEE S H, YOON H B, LEE S H. Genome-wide association study and prediction of genomic breeding values for fatty-acid composition in Korean Hanwoo cattle using a high-density single-nucleotide polymorphism array. Journal of Animal Science, 2018, 96(10): 4063-4075.
doi: 10.1093/jas/sky280
pmid: 30265318
|
| [12] |
AL KALALDEH M, GIBSON J, DUIJVESTEIJN N, DAETWYLER H D, MACLEOD I, MOGHADDAR N, LEE S H, VAN DER WERF J H J. Using imputed whole-genome sequence data to improve the accuracy of genomic prediction for parasite resistance in Australian sheep. Genetics, Selection, Evolution, 2019, 51(1): 32.
doi: 10.1186/s12711-019-0476-4
pmid: 31242855
|
| [13] |
YE S P, YUAN X L, LIN X R, GAO N, LUO Y Y, CHEN Z M, LI J Q, ZHANG X Q, ZHANG Z. Imputation from SNP chip to sequence: A case study in a Chinese indigenous chicken population. Journal of Animal Science and Biotechnology, 2018, 9: 30.
doi: 10.1186/s40104-018-0241-5
pmid: 29581880
|
| [14] |
LIU R R, XING S Y, WANG J, ZHENG M Q, CUI H X, CROOIJMANS R P M A, LI Q H, ZHAO G P, WEN J. A new chicken 55K SNP genotyping array. BMC Genomics, 2019, 20(1): 410.
doi: 10.1186/s12864-019-5736-8
pmid: 31117951
|
| [15] |
PURCELL S, NEALE B, TODD-BROWN K, THOMAS L, FERREIRA M A R, BENDER D, MALLER J, SKLAR P, DE BAKKER P I W, DALY M J, SHAM P C. PLINK: A tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics, 2007, 81(3): 559-575.
doi: 10.1086/519795
|
| [16] |
BROWNING B L, ZHOU Y, BROWNING S R. A one-penny imputed genome from next-generation reference panels. The American Journal of Human Genetics, 2018, 103(3): 338-348.
doi: 10.1016/j.ajhg.2018.07.015
|
| [17] |
STAHL K, GOLA D, KÖNIG I R. Assessment of imputation quality: Comparison of phasing and imputation algorithms in real data. Frontiers in Genetics, 2021, 12: 724037.
doi: 10.3389/fgene.2021.724037
|
| [18] |
VANRADEN P M. Efficient methods to compute genomic predictions. Journal of Dairy Science, 2008, 91(11): 4414-4423.
doi: 10.3168/jds.2007-0980
pmid: 18946147
|
| [19] |
YANG J, LEE S H, GODDARD M E, VISSCHER P M. GCTA: A tool for genome-wide complex trait analysis. The American Journal of Human Genetics, 2011, 88(1): 76-82.
doi: 10.1016/j.ajhg.2010.11.011
|
| [20] |
NI G Y, CAVERO D, FANGMANN A, ERBE M, SIMIANER H. Whole-genome sequence-based genomic prediction in laying chickens with different genomic relationship matrices to account for genetic architecture. Genetics, Selection, Evolution, 2017, 49(1): 8.
doi: 10.1186/s12711-016-0277-y
pmid: 28093063
|
| [21] |
ELBASYONI I S, LORENZ A J, GUTTIERI M, FRELS K, BAENZIGER P S, POLAND J, AKHUNOV E. A comparison between genotyping-by-sequencing and array-based scoring of SNPs for genomic prediction accuracy in winter wheat. Plant Science, 2018, 270: 123-130.
doi: S0168-9452(17)30945-7
pmid: 29576064
|
| [22] |
王家迎. 基于填充序列数据的基因组选择研究[D]. 广州: 华南农业大学, 2018.
|
|
WANG J Y. The study of genome selection by using imputed whole sequence data[D]. Guangzhou: South China Agricultural University, 2018. (in Chinese)
|
| [23] |
SU G, BRØNDUM R F, MA P, GULDBRANDTSEN B, AAMAND G P, LUND M S. Comparison of genomic predictions using medium- density (-54, 000) and high-density (-777, 000) single nucleotide polymorphism marker panels in Nordic Holstein and Red Dairy Cattle populations. Journal of Dairy Science, 2012, 95(8): 4657-4665.
doi: 10.3168/jds.2012-5379
|
| [24] |
PÉREZ-ENCISO M, RINCÓN J C, LEGARRA A. Sequence- vs. chip-assisted genomic selection: Accurate biological information is advised. Genetics, Selection, Evolution, 2015, 47(1): 43.
doi: 10.1186/s12711-015-0117-5
|
| [25] |
MANOLIO T A, COLLINS F S, COX N J, GOLDSTEIN D B, HINDORFF L A, HUNTER D J, MCCARTHY M I, RAMOS E M, CARDON L R, CHAKRAVARTI A, et al. Finding the missing heritability of complex diseases. Nature, 2009, 461(7265): 747-753.
doi: 10.1038/nature08494
|
| [26] |
HAYES B J, BOWMAN P J, DAETWYLER H D, KIJAS J W, VAN DER WERF J H J. Accuracy of genotype imputation in sheep breeds. Animal Genetics, 2012, 43(1): 72-80.
doi: 10.1111/j.1365-2052.2011.02208.x
pmid: 22221027
|
| [27] |
HICKEY J M, CROSSA J, BABU R, DE LOS CAMPOS G. Factors affecting the accuracy of genotype imputation in populations from several maize breeding programs. Crop Science, 2012, 52(2): 654-663.
doi: 10.2135/cropsci2011.07.0358
|
| [28] |
LIN P, HARTZ S M, ZHANG Z H, SACCONE S F, WANG J, TISCHFIELD J A, EDENBERG H J, KRAMER J R, M GOATE A, BIERUT L J, RICE J P. A new statistic to evaluate imputation reliability. PLoS ONE, 2010, 5(3): e9697.
doi: 10.1371/journal.pone.0009697
|
| [29] |
MA P, BRØNDUM R F, ZHANG Q, LUND M S, SU G. Comparison of different methods for imputing genome-wide marker genotypes in Swedish and Finnish Red Cattle. Journal of Dairy Science, 2013, 96(7): 4666-4677.
doi: 10.3168/jds.2012-6316
pmid: 23684022
|
| [30] |
NI G Y, STROM T M, PAUSCH H, REIMER C, PREISINGER R, SIMIANER H, ERBE M. Comparison among three variant callers and assessment of the accuracy of imputation from SNP array data to whole-genome sequence level in chicken. BMC Genomics, 2015, 16: 824.
doi: 10.1186/s12864-015-2059-2
pmid: 26486989
|







 ), 朱墨1, 陈艳茹1, 童世锋1, 赵桂苹2, 刘杨1(
), 朱墨1, 陈艳茹1, 童世锋1, 赵桂苹2, 刘杨1(