






中国农业科学 ›› 2019, Vol. 52 ›› Issue (4): 579-590.doi: 10.3864/j.issn.0578-1752.2019.04.001
• 作物遗传育种·种质资源·分子遗传学 • 下一篇
杨宇昕,邹枨( )
)
收稿日期:2018-10-30
接受日期:2018-12-09
出版日期:2019-02-16
发布日期:2019-02-27
通讯作者:
邹枨
作者简介:杨宇昕,基金资助:
YANG YuXin,ZOU Cheng()
Received:2018-10-30
Accepted:2018-12-09
Online:2019-02-16
Published:2019-02-27
Contact:
Cheng ZOU
摘要:
【目的】玉米起源于热带地区,经过自然和人工选择,广泛的种植于温带地区。开花是玉米生长发育的中心环节,也是热带玉米向温带环境种植的主要适应性性状。鉴定玉米在驯化过程中出现的受选择基因区段,并进一步挖掘开花候选基因,为玉米的群体改良、开花遗传机理解析提供数据支撑。【方法】首先单独分析30份温带玉米自交系和21份热带玉米自交系的单倍型数据,通过过滤高缺失和等位基因频率较低的变异位点,得到高质量的SNP(single nucleotide polymorphism)标记,利用SnpEff软件对温带和热带玉米群体的基因组多态性位点进行了功能预测。其次过滤得到同时存在于温带和热带玉米的高质量SNP标记,对温带和热带玉米的基因型数据进行主成分分析(principle component analysis,PCA)以确定其群体结构,之后利用群体分化指数(fixation index,FST)和群体间扩展单倍型纯合度(cross population extended haplotype homozygosity,XP-EHH)法分析温带和热带玉米群体间的选择信号分布情况,选择FST和XP-EHH值的top 1%为阈值,筛选得到受选择位点。通过对SNP进行功能注释得到温热带玉米群体受到选择的基因。利用agriGO工具对候选驯化基因进行功能富集分析。利用相关的生物信息学数据库对候选基因进行功能注释,进一步鉴定玉米驯化过程中的开花候选基因。【结果】通过对温热带玉米群体的高测序深度的SNP进行分析,发现热带玉米群体的SNP数目为14 123 408个,温带玉米群体的SNP数目为8 791 673个,鉴定到的SNP主要分布于基因间区。2个群体中均存在的SNP标记数目是204 752个。主成分分析表明温带和热带玉米可以显著的分为两个类群。FST选择信号的top 1%是0.3593,共鉴定到557个候选驯化基因,XP-EHH选择信号法的top 1%是3.2681,共鉴定到1 913个候选基因。鉴定到多个候选基因与玉米的开花调控密切相关,包括ZmCCT9、COL1、GRMZM2G387528。如ZmCCT9抑制开花基因ZCN8的表达,导致玉米在长日照环境下出现晚花表型,是一个重要的开花调控基因;COL1与开花促进因子FT蛋白互作,加速玉米开花以适应长日照环境;GRMZM2G387528的功能注释揭示该基因是一个光敏色素互作因子,与光周期基因ZmphyB1互作。【结论】热带玉米群体具有更高的遗传多态性,筛选到一系列参与了热带玉米和温带玉米的分化候选基因,并且重点挖掘了参与其中的玉米开花调控相关基因。
杨宇昕,邹枨. 基于温带和热带玉米群体全基因组FST和XP-EHH的选择信号检测[J]. 中国农业科学, 2019, 52(4): 579-590.
YANG YuXin,ZOU Cheng. Genome-Wide Detection of Selection Signal in Temperate and Tropical Maize Populations with Use of FST and XP-EHH[J]. Scientia Agricultura Sinica, 2019, 52(4): 579-590.
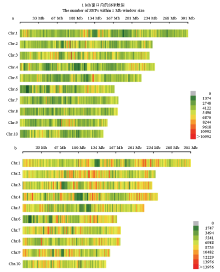
图1
温、热带玉米群体SNP在基因组上的分布 a:温带玉米群体SNP分布;b:热带玉米群体SNP分布。横轴代表染色体的物理位置,窗口大小为1 Mb区间。深绿色代表SNP密度小的区域,红色代表SNP密度高的区域"
表1
温带和热带玉米群体的SNP在基因组区间的分布比例"
| 类型 Type | 温带 Temperate | 热带 Tropical | ||
|---|---|---|---|---|
| 数目 Count | 百分比 Percent (%) | 数目 Count | 百分比 Percent (%) | |
| Downstream | 2722265 | 18.14 | 4507749 | 18.54 |
| Exon | 326417 | 2.18 | 463543 | 1.91 |
| Intergenic | 7686519 | 51.22 | 12389340 | 50.97 |
| Intron | 1257112 | 8.38 | 2046559 | 8.42 |
| Splice_Site_Acceptor | 647 | 0.00 | 1005 | 0.00 |
| Splice_Site_Donor | 602 | 0.00 | 977 | 0.00 |
| Spice_Site_Region | 28482 | 0.19 | 45454 | 0.19 |
| Upstream | 2667996 | 17.78 | 4376510 | 18.00 |
| UTR_3_Prime | 192736 | 1.28 | 297085 | 1.22 |
| UTR_5_Prime | 124412 | 0.83 | 180849 | 0.74 |

图2
温带(a)和热带(b)玉米自交系表型"

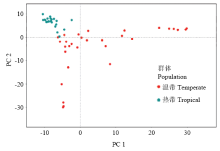
图3
温、热带群体的主成分分析"
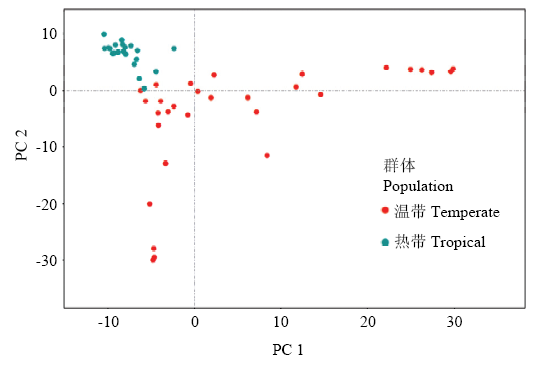
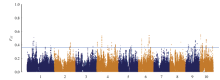
图4
全基因组水平上的FST分布 阈值线代表FST值的top 1%"
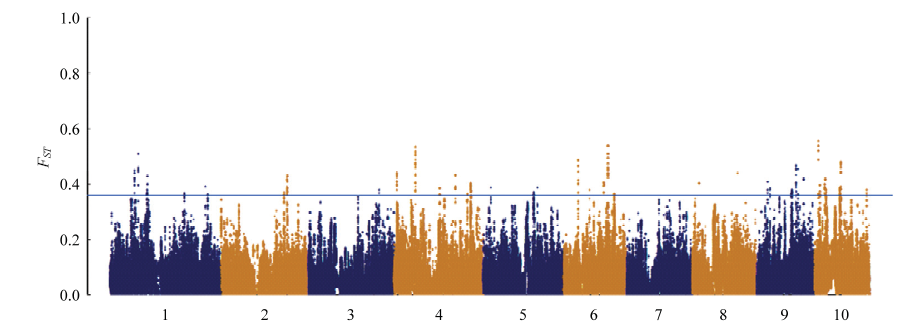
图5
温、热带玉米全基因组水平上的XP-EHH选择信号分布 阈值线代表选择信号值的top 1%"
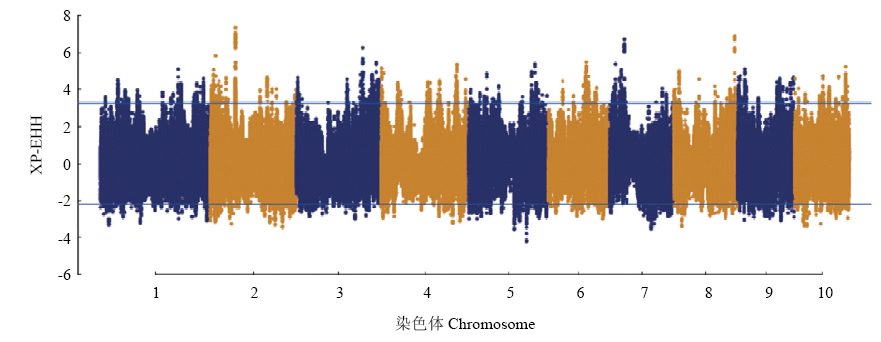
表2
FST选择信号得到的基因进行GO富集分析的结果"
| GO条目 GO term | 通路 Ontology | 描述 Description | P值 P-value | 错误发现率 FDR |
|---|---|---|---|---|
| GO:0008064 | P | 肌动蛋白聚合或解聚的调节 Regulation of actin polymerization or depolymerization | 1.30E-06 | 0.00011 |
| GO:0032271 | P | 蛋白质聚合的调节Regulation of protein polymerization | 1.30E-06 | 0.00011 |
| GO:0030832 | P | 微丝长度的调节Regulation of actin filament length | 1.30E-06 | 0.00011 |
| GO:0030833 | P | 微丝聚合反应的调节Regulation of actin filament polymerization | 1.30E-06 | 0.00011 |
| GO:0032970 | P | 肌动蛋白丝的调控过程Regulation of actin filament-based process | 1.30E-06 | 0.00011 |
| GO:0044087 | P | 细胞组分生物合成的调节Regulation of cellular component biogenesis | 1.30E-06 | 0.00011 |
| GO:0030041 | P | 肌动蛋白丝的聚合Actin filament polymerization | 2.20E-06 | 0.00014 |
| GO:0008154 | P | 激动蛋白丝的聚合或者解聚Actin polymerization or depolymerization | 2.20E-06 | 0.00014 |
| GO:0007015 | P | 肌动蛋白丝组织Actin filament organization | 2.20E-06 | 0.00014 |
| GO:0032535 | P | 细胞组分大小的调节Regulation of cellular component size | 3.10E-06 | 0.00016 |
| GO:0090066 | P | 解剖学结构大小的调节Regulation of anatomical structure size | 3.10E-06 | 0.00016 |
| GO:0033043 | P | 细胞器组织的调节Regulation of organelle organization | 4.20E-06 | 0.00021 |
| GO:0051128 | P | 细胞成分组织的调节Regulation of cellular component organization | 1.20E-05 | 0.00057 |
| GO:0030036 | P | 肌动蛋白细胞骨架组织Actin cytoskeleton organization | 0.00071 | 0.029 |
| GO:0030029 | P | 肌动蛋白丝基过程Actin filament-based process | 0.00071 | 0.029 |
| GO:0006996 | P | 细胞器组织Organelle organization | 0.0012 | 0.046 |
表3
选择信号鉴定得到的候选基因功能注释"
| 基因 Gene | 染色体 Chromosome | 物理位置 Position (Mb) | FST值a FST value | XP-EHH值b XP-EHH value | 注释c Annotation |
|---|---|---|---|---|---|
| GRMZM2G366434d | 5 | 17.336—17.338 | 0.3688** | 4.6263** | AP2-ErEBP转录因子206,ereb206 AP2-EREBP-transcription factor 206, ereb206 |
| GRMZM2G145579d | 4 | 47.618—47.620 | 0.5071** | 3.8724* | bHLH转录因子165,bhlh165 bHLH-transcription factor 165, bhlh165 |
| GRMZM2G023325 | 1 | 66.506—66.507 | 0.7845** | 氧氮杂萘酮合成12,bx12 Benzoxazinone synthesis12,bx12 | |
| GRMZM2G074094 | 1 | 199.519—199.525 | 0.4380** | SET结构域,sdg103 SET domain group 103, sdg103 | |
| GRMZM2G092091 | 1 | 220.057—220.061 | 0.4136** | bHLH转录因子27,bhlh27 bHLH-transcription factor 27,bhlh27 | |
| GRMZM2G478417 | 1 | 272.109—272.113 | 0.3706** | bZIP转录因子49,bzip49 bZIP-transcription factor 49,bzip49 | |
| GRMZM2G174834 | 4 | 159.692—159.694 | 0.3952** | WRI1转录因子,wri2 WRI1 transcription factor2, wri2 | |
| GRMZM2G126505 | 4 | 185.907—185.912 | 0.3879** | 脱落酸8'-羟化酶2,abh12 Abscisic acid 8'-hydroxylase2, abh12 | |
| GRMZM2G070523 | 5 | 20.219—20.221 | 0.3693** | MYB转录因子129,myb129 MYB-transcription factor 129, myb129 | |
| GRMZM2G004334 | 6 | 109.244—109.247 | 0.5006** | 同源框转录因子10,hb10 Homeobox-transcription factor 10, hb10 | |
| GRMZM2G170148 | 9 | 110.859—110.870 | 0.3954** | MYB相关转录因子86,mybr86 MYB-related-transcription factor 86, mybr86 | |
| GRMZM2G156013 | 10 | 141.014—141.018 | 0.4835** | 丝氨酸-苏氨酸激酶4,stk4 Serine-threonine kinase4, stk4 | |
| GRMZM2G143602 | 2 | 8.289—8.295 | 4.1470* | CK2蛋白激酶α1,cka1 CK2 protein kinase alpha 1, cka1 | |
| GRMZM2G001289 | 2 | 15.033—15.035 | 3.7898* | 同源框转录因子75,hb75 Homeobox-transcription factor 75, hb75 | |
| GRMZM2G102059 | 2 | 236.711—236.716 | 3.6779* | ABI3-VP1转录因子12,abi12 ABI3-VP1-transcription factor 12, abi12 | |
| GRMZM2G089638 | 3 | 1.464—1.469 | 4.1762* | TCP转录因子31,tcptf31 TCP-transcription factor 31, tcptf31 | |
| GRMZM2G087804 | 3 | 140.859—140.861 | 3.9809* | 金色植物2,g2 Golden plant2, g2 | |
| GRMZM2G015875 | 4 | 47.618—47.620 | 4.5721* | NMCP/CRWN同源因子1,nch1 NMCP/CRWN-Homologous1, nch1 | |
| GRMZM2G145579 | 4 | 161.101—161.108 | 3.8724* | bHLH转录因子165,bhlh165 bHLH-transcription factor 165, bhlh165 | |
| GRMZM5G880069 | 5 | 184.337—184.342 | 3.8136* | WRKY转录因子109,wrky109 WRKY-transcription factor 109, wrky109 | |
| GRMZM2G129783 | 6 | 106.637—106.640 | 3.7924* | 五肽重复蛋白346,ppr346 Pentatricopeptide repeat protein346, ppr346 | |
| GRMZM2G387528 | 8 | 0.935—0.938 | 4.2271* | 光敏色素作用因子3,pif3 Phytochrome interacting factor3, pif3 | |
| GRMZM2G028054 | 8 | 170.725—170.728 | 3.7593* | MYB转录因子74,myb74 MYB-transcription factor 74, myb74 | |
| GRMZM2G009808 | 9 | 107.641—107.645 | 5.3817** | 乌头酸梅3,aco30 Aconitase3, aco30 | |
| GRMZM2G139082 | 9 | 116.292—116.294 | 4.3570* | 热冲击互补因子1,hscf1 Heat shock complementing factor1, hscf1 | |
| GRMZM2G013671 | 9 | 152.030—152.038 | 3.7187* | β扩增蛋白5 Beta expansin5, expb5 | |
| GRMZM2G474769 | 10 | 78.160—78.161 | 4.8528** | 下胚轴伸长蛋白1,lhy1 Late hypocotyl elongation protein ortholog1, lhy1 | |
| GRMZM2G180168 | 10 | 78.333—78.337 | 4.6254** | ABI3-VP1转录因子23,abi23 ABI3-VP1-transcription factor 23, abi23 |
| [1] |
PIPERNO D R, RANERE A J, HOLST I, IRIARTE J, DICKAU R . Starch grain and phytolith evidence for early ninth millennium BP maize from the Central Balsas River Valley, Mexico. Proceedings of the National Academy of Sciences of the United States of America, 2009,106(13):5019-5024.
doi: 10.1073/pnas.0812525106 pmid: 19307570 |
| [2] |
MATSUOKA Y, VIGOUROUX Y, GOODMAN M M, SANCHEZ J, BUCKLER E, DOEBLEY J . A single domestication for maize shown by multilocus microsatellite genotyping. Proceedings of the National Academy of Sciences of the United States of America, 2002,99(9):6080-6084.
doi: 10.1073/pnas.052125199 pmid: 11983901 |
| [3] |
VAN HEERWAARDEN J, DOEBLEY J, BRIGGS W H, GLAUBITZ J C, GOODMAN M M, GONZALEZ J D J S, ROSS-IBARRA J . Genetic signals of origin, spread, and introgression in a large sample of maize landraces. Proceedings of the National Academy of Sciences of the United States of America, 2011,108(3):1088-1092.
doi: 10.1073/pnas.1013011108 pmid: 21189301 |
| [4] |
BUCKLER E S, HOLLAND J B, BRADBURY P J, ACHARYA C B, BROWN P J, BROWNE C, ERSOZ E, FLINT-GARCIA S, GARCIA A, GLAUBITZ J C , et at. The genetic architecture of maize flowering time. Science, 2009,325(5941):714-718.
doi: 10.1126/science.1174276 |
| [5] |
SWARTS K, GUTAKER R M, BENZ B, BLAKE M, BUKOWSKI R, HOLLAND J, KRUSE-PEEPLES M, LEPAK N, PRIM L, ROMAY M C , et at. Genomic estimation of complex traits reveals ancient maize adaptation to temperate North America. Science, 2017,357(6350):512-515.
doi: 10.1126/science.aam9425 pmid: 28774930 |
| [6] |
LU Y L, YAN J B, GUIMARAES C T, TABA S, HAO Z F, GAO S B, CHEN S J, LI J S, ZHANG S H, VIVEK B S , et at. Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theoretical and Applied Genetics, 2009,120(1):93-115.
doi: 10.1007/s00122-009-1162-7 pmid: 19823800 |
| [7] |
TALLURY S, GOODMAN M . Experimental evaluation of the potential of tropical germplasm for temperate maize improvement. Theoretical and Applied Genetics, 1999,98(1):54-61.
doi: 10.1007/s001220051039 |
| [8] |
HALLAUER A R, CARENA M J . Adaptation of tropical maize germplasm to temperate environments. Euphytica, 2013,196(1):1-11.
doi: 10.1007/s10681-013-1017-9 |
| [9] |
KIMURA M . Evolutionary rate at the molecular level. Nature, 1968,217(5129):624-626.
doi: 10.1038/217624a0 |
| [10] |
SMITH J M, HAIGH J . The hitch-hiking effect of a favourable gene. Genetics Research, 1974,23(1):23-35.
doi: 10.1017/S0016672300014634 pmid: 4407212 |
| [11] |
TAJIMA F . Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 1989,123(3):585-595.
doi: 10.1101/gad.3.11.1801 pmid: 2513255 |
| [12] |
NIELSEN R, WILLIAMSON S, KIM Y, HUBISZ M J, CLARK A G, BUSTAMANTE C . Genomic scans for selective sweeps using SNP data. Genome Research, 2005,15(11):1566-1575.
doi: 10.1101/gr.4252305 pmid: 16251466 |
| [13] |
SABETI P C, REICH D E, HIGGINS J M, LEVINE H Z, RICHTER D J, SCHAFFNER S F, GABRIEL S B, PLATKO J V, PATTERSON N J, MCDONALD G J , et at. Detecting recent positive selection in the human genome from haplotype structure. Nature, 2002,419(6909):832.
doi: 10.1038/nature01140 pmid: 12397357 |
| [14] |
VOIGHT B F, KUDARAVALLI S, WEN X Q, PRITCHARD J K . A map of recent positive selection in the human genome. PLoS Biology, 2006,4(3):e72.
doi: 10.1371/journal.pbio.0040072 pmid: 1892825 |
| [15] |
SABETI P C, VARILLY P, FRY B, LOHMUELLER J, HOSTETTER E, COTSAPAS C, XIE X, BYRNE E H, MCCARROLL S A , et at. Genome-wide detection and characterization of positive selection in human populations. Nature, 2007,449(7164):913.
doi: 10.1038/nature06250 |
| [16] |
WRIGHT S . The genetical structure of populations. Annals of Eugenics, 1949,15(1):323-354.
doi: 10.1007/978-3-642-88415-3 |
| [17] |
WEIR B S, COCKERHAM C C . Estimating F-statistics for the analysis of population structure. Evolution, 1984,38(6):1358-1370.
doi: 10.1111/j.1558-5646.1984.tb05657.x pmid: 28563791 |
| [18] |
GIANOLA D, SIMIANER H, QANBARI S . A two-step method for detecting selection signatures using genetic markers. Genetics Research, 2010,92(2):141-155.
doi: 10.1017/S0016672310000121 pmid: 20515517 |
| [19] |
AXELSSON E, RATNAKUMAR A, ARENDT M L, MAQBOOL K, WEBSTER M T, Perloski M, Liberg O, ARNEMO J M, HEDHAMMAR A, LINDBLAD-TOH K . The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature, 2013,495(7441):360.
doi: 10.1038/nature11837 |
| [20] |
LIU K J, GOODMAN M, MUSE S, SMITH J S, BUCKLER E, DOEBLEY J . Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics, 2003,165(4):2117-2128.
doi: 10.1017/S001667230006426 pmid: 14704191 |
| [21] |
HE C, FU J J, ZHANG J, Li Y X, ZHENG J, ZHANG H W, YANG X H, WANG J H, WANG G Y . A gene-oriented haplotype comparison reveals recently selected genomic regions in temperate and tropical maize germplasm. PLoS ONE, 2017,12(1):e0169806.
doi: 10.1371/journal.pone.0169806 pmid: 5242465 |
| [22] |
SCHNABLE P S, WARE D, FULTON R S, STEIN J C, WEI F S, PASTERNAK S, LIANG C Z, ZHANG J W, FULTON L, GRAVES T A , et at. The B73 maize genome: complexity, diversity, and dynamics. Science, 2009,326(5956):1112-1115.
doi: 10.1126/science.1178534 |
| [23] |
JIAO Y P, PELUSO P, SHI J H, LIANG T, STITZER M C, WANG B, CAMPBELL M S, STEIN J C, WEI X H, CHIN C S , et at. Improved maize reference genome with single-molecule technologies. Nature, 2017,546(7659):524.
doi: 10.1038/nature22971 pmid: 28605751 |
| [24] |
CHIA J M, SONG C, BRADBURY P J, COSTICH D, DE LEON N, DOEBLEY J, ELSHIRE R J, GAUT B, GELLER L, GLAUBITZ J C , et at. Maize HapMap2 identifies extant variation from a genome in flux. Nature Genetics, 2012,44(7):803.
doi: 10.1038/ng.2313 |
| [25] |
BUKOWSKI R, GUO X S, LU Y L, ZOU C, HE B, RONG Z Q, WANG B, XU D W, YANG B C, XIE C X , et at. Construction of the third-generation Zea mays haplotype map. GigaScience, 2017, 7(4): gix134.
doi: 10.1093/gigascience/gix134 pmid: 29300887 |
| [26] |
DANECEK P, AUTON A, ABECASIS G, ALBERS C A, BANKS E, DEPRISTO M A, HANDSAKER R E, LUNTER G, MARTH G T, SHERRY S T , et at. 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics, 2011,27(15):2156-2158.
doi: 10.1093/bioinformatics/btr330 pmid: 21653522 |
| [27] |
CINGOLANI P, PLATTS A, WANG L L, COON M, NGUYEN T, WANG L A, LAND S J, LU X Y, RUDEN D M . A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of drosophila melanogaster strain w1118; iso-2; iso-3. Fly, 2012,6(2):80-92.
doi: 10.4161/fly.19695 |
| [28] |
MA Y, DING X, QANBARI S, WEIGEND S, ZHANG Q, SIMIANER H . Properties of different selection signature statistics and a new strategy for combining them. Heredity, 2015,115(5):426.
doi: 10.1038/hdy.2015.42 pmid: 4611237 |
| [29] | HOAGLIN D C, MOSTELLER F, TUKEY J W . Understanding Robust and Exploratory Data Analysis. New York: John Wiley & Sons, 1983. |
| [30] |
SZPIECH Z A, HERNANDEZ R D . selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Molecular Biology and Evolution, 2014,31(10):2824-2827.
doi: 10.1093/molbev/msu211 pmid: 4166924 |
| [31] |
QUINLAN A R, HALL I M . BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 2010,26(6):841-842.
doi: 10.1093/bioinformatics/btq033 pmid: 20110278 |
| [32] |
TIAN T, LIU Y, YAN H Y, YOU Q, YI X, DU Z, XU W Y, SU Z . AgriGO v2. 0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Research, 2017,45(W1):W122-W129.
doi: 10.1093/nar/gkx382 pmid: 28472432 |
| [33] |
HUANG C, SUN H Y, XU D Y, LIANG Y M, WANG X F, XU G H, TIAN J G, WANG C L, LI D, WU L H , et at. ZmCCT9 enhances maize adaptation to higher latitudes. Proceedings of the National Academy of Sciences of the United States of America, 2018,115(2):E334-E341.
doi: 10.1073/pnas.1718058115 pmid: 29279404 |
| [34] |
KHAN S, ROWE S C, HARMON F G . Coordination of the maize transcriptome by a conserved circadian clock. BMC Plant Biology, 2010,10(1):126.
doi: 10.1186/1471-2229-10-126 pmid: 20576144 |
| [35] | GUO L, WANG X, ZHAO M, HUANG C, LI C, LI D, YANG C, YORK A M, XUE W, XU G, LIANG Y, CHEN Q, DOEBLEY J F, TIAN F . Stepwise cis-regulatory changes in ZCN8 contribute to maize flowering-time adaptation. Current Biology, 2018,28(18):3005-3015. |
| [36] | MENG X, MUSZYNSKI M G, DANILEVSKAYA O N . The FT-like ZCN8 gene functions as a floral activator and is involved in photoperiod sensitivity in maize. The Plant Cell, 2011,23(3):942-960. |
| [37] |
SHEEHAN M J, NACHMAN M W . Morphological and population genomic evidence that human faces have evolved to signal individual identity. Nature Communications, 2014,5:4800.
doi: 10.1038/ncomms5800 pmid: 25226282 |
| [38] |
曾滔, 赵福平, 王光凯, 吴明明, 魏彩虹, 张莉, 李利, 张红平, 杜立新 . 基于群体分化指数FST的绵羊全基因组选择信号检测. 畜牧兽医学报, 2013,44(12):1891-1899.
doi: 10.11843/j.issn.0366-6964.2013.12.005 |
|
ZENG T, ZHAO F P, WANG G K, WU M M, WEI C H, ZHANG L, LI L, ZHANG H P, DU L X . Genome-wide detection of selection signatures in sheep populations with use of population differentiation index FST. Acta Veterinaria et Zootechnica Sinica, 2013,44(12):1891-1899. (in Chinese)
doi: 10.11843/j.issn.0366-6964.2013.12.005 |
|
| [39] |
HUANG X, SANG T, ZHAO Q, QI F, ZHAO Y, LI C Y ZHU C R, LU T T, ZHANG Z W, LI M , et at. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genetics, 2010,42(11):961.
doi: 10.1038/ng.695 pmid: 20972439 |
| [40] |
MCVICKER G, GORDON D, DAVIS C, GREEN P . Widespread genomic signatures of natural selection in hominid evolution. PLoS Genetics, 2009,5(5):e1000471.
doi: 10.1371/journal.pgen.1000471 pmid: 19424416 |
| [41] | 薛周舣源, 宋显威, 吴林慧, 王露珍, 崔家安, 孙章健, 张政, 马云龙 . 畜禽选择信号检测方法及其统计学问题. 畜牧兽医学报, 2018,49(6):1099-1107. |
| XUE Z Y Y, SONG X W, WU L H, WANG L Z, CUI J A, SUN Z J, ZHANG Z, MA Y L . The identification methods of selection signatures in livestock and its statistical problems. Acta Veterinaria et Zootechnica Sinica, 2018,49(6):1099-1107. (in Chinese) | |
| [42] |
KUMAR I, SWAMINATHAN K, HUDSON K, HUDSON M E . Evolutionary divergence of phytochrome protein function in Zea mays PIF3 signaling. Journal of Experimental Botany, 2016,67(14):4231-4240.
doi: 10.1093/jxb/erw217 pmid: 27262126 |
| [1] | 柴海燕,贾娇,白雪,孟玲敏,张伟,金嵘,吴宏斌,苏前富. 吉林省玉米穗腐病致病镰孢菌的鉴定与部分菌株对杀菌剂的敏感性[J]. 中国农业科学, 2023, 56(1): 64-78. |
| [2] | 赵政鑫,王晓云,田雅洁,王锐,彭青,蔡焕杰. 未来气候条件下秸秆还田和氮肥种类对夏玉米产量及土壤氨挥发的影响[J]. 中国农业科学, 2023, 56(1): 104-117. |
| [3] | 李周帅,董远,李婷,冯志前,段迎新,杨明羡,徐淑兔,张兴华,薛吉全. 基于杂交种群体的玉米产量及其配合力的全基因组关联分析[J]. 中国农业科学, 2022, 55(9): 1695-1709. |
| [4] | 熊伟仡,徐开未,刘明鹏,肖华,裴丽珍,彭丹丹,陈远学. 不同氮用量对四川春玉米光合特性、氮利用效率及产量的影响[J]. 中国农业科学, 2022, 55(9): 1735-1748. |
| [5] | 李易玲,彭西红,陈平,杜青,任俊波,杨雪丽,雷鹿,雍太文,杨文钰. 减量施氮对套作玉米大豆叶片持绿、光合特性和系统产量的影响[J]. 中国农业科学, 2022, 55(9): 1749-1762. |
| [6] | 马小艳,杨瑜,黄冬琳,王朝辉,高亚军,李永刚,吕辉. 小麦化肥减施与不同轮作方式的周年养分平衡及经济效益分析[J]. 中国农业科学, 2022, 55(8): 1589-1603. |
| [7] | 李前,秦裕波,尹彩侠,孔丽丽,王蒙,侯云鹏,孙博,赵胤凯,徐晨,刘志全. 滴灌施肥模式对玉米产量、养分吸收及经济效益的影响[J]. 中国农业科学, 2022, 55(8): 1604-1616. |
| [8] | 张家桦,杨恒山,张玉芹,李从锋,张瑞富,邰继承,周阳晨. 不同滴灌模式对东北春播玉米籽粒淀粉积累及淀粉相关酶活性的影响[J]. 中国农业科学, 2022, 55(7): 1332-1345. |
| [9] | 谭先明,张佳伟,王仲林,谌俊旭,杨峰,杨文钰. 基于PLS的不同水氮条件下带状套作玉米产量预测[J]. 中国农业科学, 2022, 55(6): 1127-1138. |
| [10] | 冯宣军, 潘立腾, 熊浩, 汪青军, 李静威, 张雪梅, 胡尔良, 林海建, 郑洪建, 卢艳丽. 南方地区120份甜、糯玉米自交系重要目标性状和育种潜力分析[J]. 中国农业科学, 2022, 55(5): 856-873. |
| [11] | 刘苗,刘朋召,师祖姣,王小利,王瑞,李军. 氮磷配施下夏玉米临界氮浓度稀释曲线的构建与氮营养诊断[J]. 中国农业科学, 2022, 55(5): 932-947. |
| [12] | 乔远,杨欢,雒金麟,汪思娴,梁蓝月,陈新平,张务帅. 西北地区玉米生产投入及生态环境风险评价[J]. 中国农业科学, 2022, 55(5): 962-976. |
| [13] | 黄兆福, 李璐璐, 侯梁宇, 高尚, 明博, 谢瑞芝, 侯鹏, 王克如, 薛军, 李少昆. 不同种植区玉米生理成熟后田间站秆脱水的积温需求[J]. 中国农业科学, 2022, 55(4): 680-691. |
| [14] | 石习, 宁丽华, 葛敏, 邬奇, 赵涵. 玉米氮状况相关生物标记物的筛选和应用[J]. 中国农业科学, 2022, 55(3): 438-450. |
| [15] | 张建军, 党翼, 赵刚, 王磊, 樊廷录, 李尚中. 覆膜时期和施氮量对陇东旱塬玉米产量和水氮利用效率的影响[J]. 中国农业科学, 2022, 55(3): 479-490. |
|
||