






中国农业科学 ›› 2025, Vol. 58 ›› Issue (21): 4528-4543.doi: 10.3864/j.issn.0578-1752.2025.21.020
所属专题: 专题——苜蓿耐盐碱抗旱基因挖掘与育种
穆赢通( ), 路景诗, 张雨桐, 石凤翎()
), 路景诗, 张雨桐, 石凤翎()
收稿日期:2024-12-31
接受日期:2025-08-19
出版日期:2025-11-01
发布日期:2025-11-06
通信作者:
联系方式:
穆赢通,E-mail:2911426494@qq.com。
基金资助:
MU YingTong(), LU JingShi, ZHANG YuTong, SHI FengLing()
Received:2024-12-31
Accepted:2025-08-19
Published:2025-11-01
Online:2025-11-06
摘要:
【背景】干旱胁迫是限制全球农业生产力的主要非生物因子之一。系统解析植物在干旱及复水过程中的转录调控机制,对于提升作物抗旱性及实现分子育种具有重要理论与实践意义。花苜蓿作为多年生豆科牧草,具有优良的生态适应能力和抗旱潜力。【目的】通过基于转录组分析与共表达网络构建,识别直立型花苜蓿对干旱胁迫及复水响应的关键调控模块及核心功能基因,解析其潜在分子机制。【方法】设置4个处理阶段,包括正常水分处理(A组)、干旱胁迫中期(B组)、干旱胁迫后期(C组)和复水处理阶段(D组),以模拟花苜蓿在干旱-复水过程中的生理响应状态。利用高通量转录组测序获取基因表达数据,结合加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)构建基因共表达模块。通过主成分分析(principal component analysis, PCA)和KEGG通路富集分析揭示基因表达的变异趋势与功能富集规律,进一步筛选与抗旱性密切相关的模块及核心基因。最终选取MEmagenta与MEdarkgreen模块中的6个核心基因进行qRT-PCR验证,以评估转录组数据的表达一致性与模块的生物学可信度。【结果】PCA分析显示,不同处理组样本在PC1与PC2维度上明显分离,表明干旱及复水处理对花苜蓿基因表达具有阶段性影响。差异表达分析结果显示,干旱中期(B组)诱导的上调与下调基因数量最多,反映出植物在早期迅速激活应答机制;干旱后期(C组)差异表达数量有所下降,但脂肪酸降解、糖代谢等代谢路径显著富集,提示植物向稳态调控方向转变;复水阶段(D组)大部分基因表达水平趋于恢复,部分信号转导和防御通路仍保持活跃状态,反映其持续的调节能力。WGCNA分析识别出4个与处理条件显著相关的模块(|r| > 0.6),其中MEdarkgreen模块(r = 0.93)在干旱后期高表达,富集于MAPK信号通路、内质网应激、脂质代谢与黄酮类合成等路径。模块核心基因BZIP17与IRE1B分别调控蛋白质折叠、转录重编程及内质网稳态,可能在长期胁迫适应中发挥重要作用。MEmagenta模块(r = 0.82)在正常水分状态下高度表达,富集于ABA与JA信号通路、黄酮代谢等路径,核心基因ABF4与MYC2分别参与气孔调控与次生代谢调节,是干旱应答启动阶段的重要调控因子。此外,NAC072、CLPD等基因亦参与叶绿体蛋白稳态与活性氧清除等过程,进一步增强植物的胁迫缓冲能力。KEGG功能富集结果与模块功能保持高度一致性,验证了模块生物学注释的可靠性。qRT-PCR验证结果表明,所选6个核心基因在干旱中后期(B、C组)显著上调,复水后(D组)表达普遍下降,整体表达趋势与转录组数据一致,进一步支持模块识别与功能预测的准确性。【结论】本研究揭示了花苜蓿在干旱胁迫及复水过程中转录水平的动态调控特征。通过共表达网络分析,识别出两个与抗旱适应性密切相关的关键模块MEmagenta与MEdarkgreen,筛选得到ABF4、MYC2、BZIP17和IRE1B等关键调控因子。这些基因在信号转导、代谢调控与胁迫适应中发挥核心作用,代表了花苜蓿在应对干旱过程中的潜在分子机制。研究为进一步解析牧草的抗旱分子基础及其分子育种提供了理论支撑与候选靶标。
穆赢通, 路景诗, 张雨桐, 石凤翎. 基于转录组和WGCNA的直立型花苜蓿抗旱关键基因识别[J]. 中国农业科学, 2025, 58(21): 4528-4543.
MU YingTong, LU JingShi, ZHANG YuTong, SHI FengLing. Identification of Key Drought-Responsive Genes in Upright Medicago ruthenica Sojak cv. Zhilixing Based on Transcriptome Sequencing and WGCNA[J]. Scientia Agricultura Sinica, 2025, 58(21): 4528-4543.
表1
转录本注释情况"
| 数据库Database | 注释到基因数Number of genes annotated | 占比Proportion (%) |
|---|---|---|
| Kyoto Encyclopedia of Genes and Genomes (KEGG) | 20123 | 73.18 |
| Gene Ontology (GO) | 16526 | 60.10 |
| NCBI non-redundant protein sequences (Nr) | 22495 | 81.81 |
| Swiss-Prot Protein Sequence Database (SwissProt) | 14939 | 54.33 |
| Translated EMBL Nucleotide Sequence Data Library (TrEMBL) | 15227 | 55.37 |
| Eukaryotic Orthologous Groups (KOG) | 15034 | 54.67 |
| Protein Families Database (Pfam) | 16390 | 59.60 |
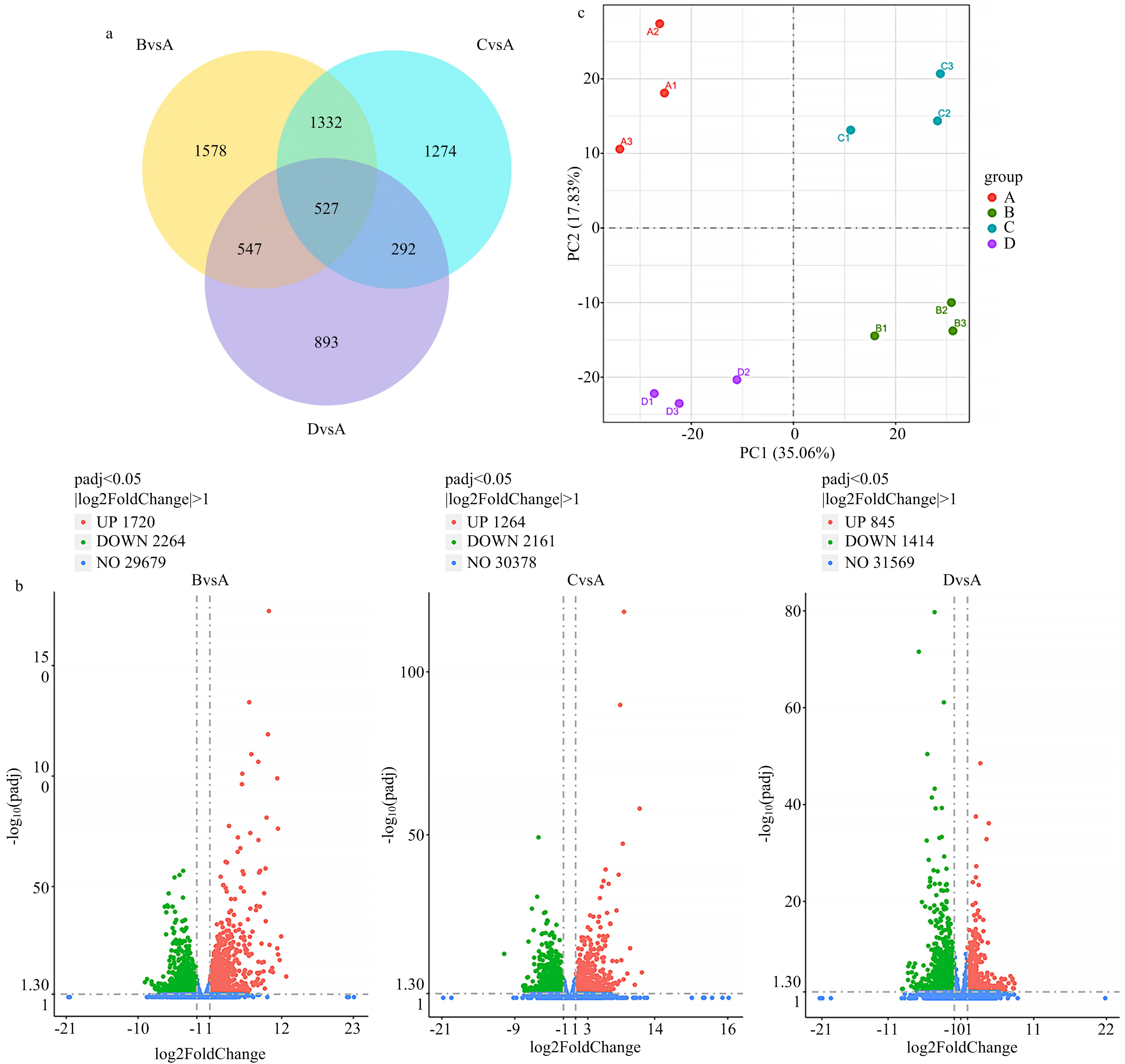
图1
干旱胁迫与复水处理下差异表达基因的总体分析 a:不同处理组与对照组(BvsA、CvsA、DvsA)差异基因的韦恩图交集关系;b:三组比较中差异表达基因的火山图,展示显著上调、下调及非显著基因的分布情况;c:PCA二维分布图,反映各处理样本在主成分空间中的表达分离趋势"

图2
KEGG通路富集分析"

图3
软阈值的网络拓扑分析、基因聚类树和模块分割 a:软阈值筛选图;b:基因聚类树图"

图4
模块与样品处理条件的相关性热图"

图5
关键基因模块 KEGG 富集分析 a:MEmagenta模块;b:MEdarkgreen模块 a: MEmagenta module; b: MEdarkgreen module"

图6
基因互作网络图 a:MEmagenta核心基因互作网络图; b:MEmagenta核心基因表达;c:MEdarkgreen核心基因互作网络图; d:MEdarkgreen核心基因表达"
表2
核心基因功能注释表"
| 模块 Module | 核心基因 Hub gene | 核心基因在拟南 芥的同源基因 Hub gene in A. thaliana | log2FoldChange (BvsA) | 基因功能 Gene function |
|---|---|---|---|---|
| MEmagenta | ABF4 | AT4G34000.1 | 1.58 | 参与干旱胁迫下的ABA信号传导通路,调控下游防御基因的表达,增强植物的抗逆性 Involved in ABA signaling pathways under drought stress, regulating the expression of downstream defense genes and enhancing plant stress tolerance |
| CLPD | AT1G06850.1 | 2.22 | ATP依赖型蛋白酶,主要作用于叶绿体中蛋白质的降解与稳态调节,维持光合作用正常进行 ATP-dependent protease primarily functioning in chloroplast protein degradation and homeostasis, ensuring the proper function of photosynthesis | |
| LTI65 | AT5G52310.1 | 0.53 | 低温诱导基因,参与植物低温适应过程,调控冷胁迫下的细胞膜稳定性 Cold-inducible gene involved in plant cold adaptation, regulating cell membrane stability under cold stress | |
| NAC072 | AT4G27410.1 | 1.37 | NAC转录因子,调控植物细胞死亡和胁迫响应基因的表达,与植物防御机制密切相关 NAC transcription factor regulating programmed cell death and the expression of stress-responsive genes, closely associated with plant defense mechanisms | |
| MYC2 | AT1G32640.1 | 0.49 | bHLH转录因子,调控茉莉酸信号通路相关基因的表达,在胁迫响应和次生代谢调控中发挥重要作用 bHLH transcription factor regulating jasmonic acid pathway-related genes, playing a critical role in stress responses and secondary metabolism | |
| PXG3 | AT5G15120.1 | 3.74 | 参与植物体内的过氧化物分解,与植物抗氧化应激机制相关,调节细胞内活性氧水平 Involved in peroxidation decomposition within plants, associated with antioxidant stress mechanisms and regulating intracellular reactive oxygen species levels | |
| NCED3 | AT3G14440.1 | 0.41 | 胡萝卜素裂解酶,催化ABA合成的关键步骤,调控植物在干旱胁迫下的抗逆性 Carotenoid cleavage dioxygenase catalyzing a key step in ABA biosynthesis, regulating plant drought stress tolerance | |
| MEdarkgreen | BZIP17 | AT2G40950.1 | 0.79 | bZIP转录因子,调控植物在盐胁迫和内质网应激条件下的基因表达,参与胁迫信号传导网络 bZIP transcription factor regulating gene expression under salt and endoplasmic reticulum stress, involved in stress signaling networks |
| JUF2 | AT3G61260.1 | 3.59 | 可能调控细胞分裂素相关基因的表达,与细胞分裂与增殖密切相关 Potentially regulates cytokinin-related gene expression, closely associated with cell division and proliferation | |
| IRE1B | AT5G24360.1 | 0.85 | 内质网应激信号通路的重要因子,裂解XBP1 mRNA以激活下游应激基因表达 Key factor in endoplasmic reticulum stress signaling, splicing XBP1 mRNA to activate downstream stress-responsive genes | |
| S2P | AT1G20130.1 | 0.36 | 跨膜蛋白裂解酶,调控信号蛋白的活性,与内质网-细胞核信号转导过程相关 Membrane-bound protease regulating the activity of signal proteins, associated with endoplasmic reticulum-to-nucleus signal transduction | |
| BIP3 | AT1G09080.1 | 0.63 | 内质网伴侣蛋白,帮助蛋白质正确折叠,缓解内质网压力,参与内质网应激响应 Endoplasmic reticulum chaperone protein assisting proper protein folding, alleviating ER stress, and participating in ER stress responses | |
| SPC25 | AT3G11980.1 | 1.47 | 动态微管蛋白复合体的核心蛋白,调控细胞分裂过程中纺锤体的组装与稳定性 Core protein of the dynamic microtubule complex, regulating spindle assembly and stability during cell division | |
| BZIP9 | AT5G24800.1 | 1.37 | bZIP转录因子,与植物激素响应和代谢相关基因的调控密切相关 bZIP transcription factor closely involved in regulating genes related to plant hormone responses and metabolism | |
| IRE1A | AT2G17520.1 | 0,85 | 内质网应激通路的调控因子,功能类似IRE1B,增强植物对内质网压力的适应能力 Regulatory factor of the ER stress pathway, functionally similar to IRE1B, enhancing plant adaptation to ER stress | |
| BZIP8 | AT4G34590.1 | 0.48 | bZIP转录因子,可能参与光合产物分配与胁迫信号的调控过程 bZIP transcription factor potentially involved in the allocation of photosynthetic products and stress signaling | |
| SBT6.1 | AT1G32940.1 | 0.09 | 丝氨酸蛋白酶,可能参与胁迫下细胞壁的重塑过程,调控细胞生长与分化 Serine protease potentially involved in cell wall remodeling under stress, regulating cell growth and differentiation | |
| BZIP10 | AT4G02640.1 | 0.41 | bZIP转录因子,可能与光信号通路相关,调控光合作用和植物生长发育基因的表达 bZIP transcription factor potentially associated with light signaling pathways, regulating photosynthesis and genes related to plant growth and development |
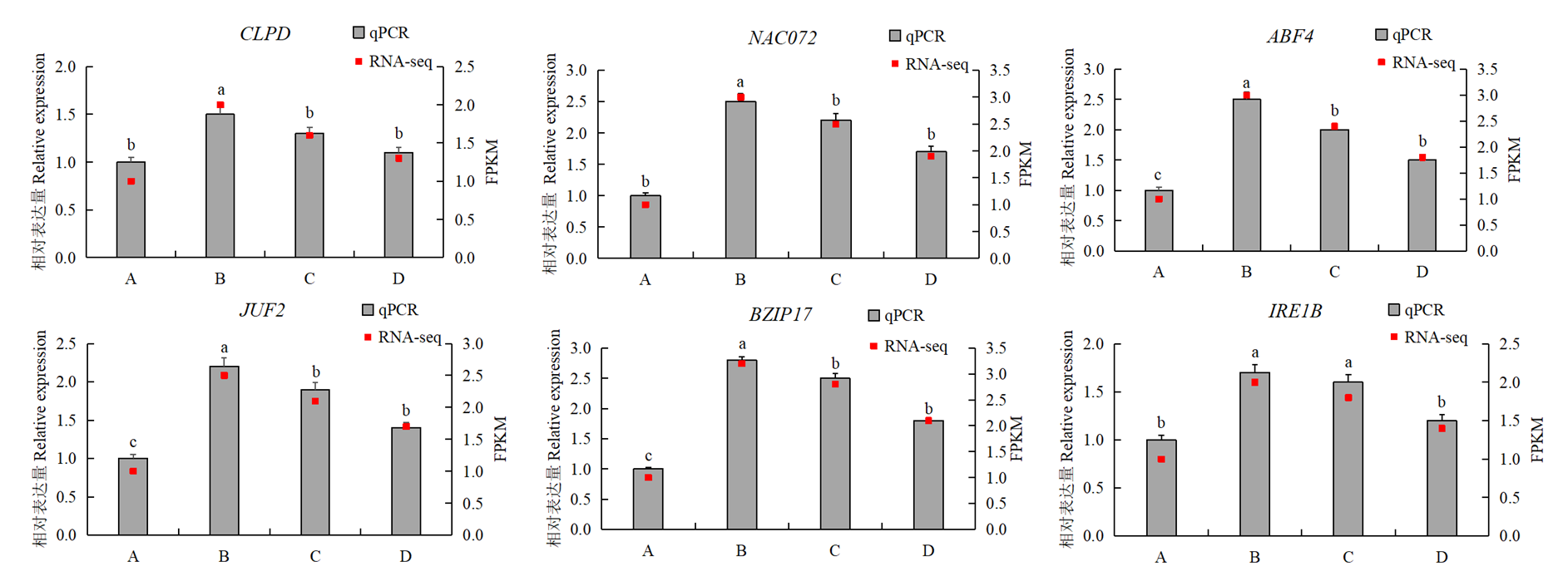
图7
qRT-PCR验证"
| [1] |
|
| [2] |
|
| [3] |
doi: 10.1007/s00018-014-1767-0 pmid: 25336153 |
| [4] |
doi: 10.1016/S1672-6308(14)60289-4 |
| [5] |
|
| [6] |
|
| [7] |
杜宝红, 石凤翎, 王晓英. 扁蓿豆根系及根颈形态变化与抗寒性关系的初步研究. 农业与技术, 2018, 38(18): 17-18.
|
|
|
|
| [8] |
吴征镒. 中国植物志. 北京: 科学出版社, 1998: 318-320.
|
|
|
|
| [9] |
|
| [10] |
刘洁, 胡蝶, 楚海家, 闫娟, 李建强. 花苜蓿抗旱耐盐EST-SSR标记筛选. 植物科学学报, 2013, 31(5): 493-499.
|
|
|
|
| [11] |
董奇琦, 艾鑫, 张艳正, 张克朝, 周东英, 王晓光, 蒋春姬, 赵姝丽, 钟超, 王婧, 于海秋, 赵新华. 干旱胁迫对不同耐性花生品种生理特性及产量的影响. 沈阳农业大学学报, 2020, 51(1): 18-26.
|
|
|
|
| [12] |
李欢, 樊军锋, 高建社, 周永学. 黑杨叶片旱生结构的比较. 西北林学院学报, 2013, 28(3): 113-118.
|
|
|
|
| [13] |
pmid: 23029918 |
| [14] |
肖红, 王芳, 段春华, 景媛媛, 陈陆军, 张建文, 杨海磊, 鱼小军. 不同扁蓿豆种质孕蕾期抗旱性综合评价. 干旱地区农业研究, 2016, 34(5): 62-68.
|
|
|
|
| [15] |
doi: 10.1186/1471-2105-9-559 pmid: 19114008 |
| [16] |
高晨曦, 郝陆洋, 胡悦, 李永祥, 张登峰, 李春辉, 宋燕春, 石云素, 王天宇, 黎裕, 刘旭洋. 干旱条件下玉米转座子插入关联的表观调控分析. 中国农业科学, 2024, 57(6): 1034-1049. doi: 10.3864/j.issn.0578-1752.2024.06.002.
|
|
|
|
| [17] |
陈晓涓, 王海菊, 王富敏, 雍清青, 黄顺满, 屈燕. 基于WGCNA鉴定全缘叶绿绒蒿类黄酮合成途径关键基因. 中国农业科学, 2024, 57(15): 3053-3070. doi: 10.3864/j.issn.0578-1752.2024.15.011.
|
|
|
|
| [18] |
彭佳伟, 张叶, 寇单单, 杨丽, 刘晓飞, 张学英, 陈海江, 田义. ‘仓方早生’桃及其早熟芽变不同发育时期果实的转录组分析. 中国农业科学, 2023, 56(5): 964-980. doi: 10.3864/j.issn.0578-1752.2023.05.012.
|
|
|
|
| [19] |
doi: 10.1080/10543400903572753 pmid: 20309759 |
| [20] |
黄涛, 熊辉岩, 段瑞君. 大麦干旱胁迫多转录组整合分析及共表达网络的构建. 植物生理学报, 2024, 60(4): 697-713.
|
|
|
|
| [21] |
李佩婷, 赵振丽, 黄潮华, 黄国强, 徐良年, 邓祖湖, 张玉, 赵新旺. 基于转录组及WGCNA的甘蔗干旱响应调控网络分析. 作物学报, 2022, 48(7): 1583-1611.
doi: 10.3724/SP.J.1006.2022.14121 |
|
|
|
| [22] |
秦天元, 孙超, 毕真真, 梁文君, 李鹏程, 张俊莲, 白江平. 基于WGCNA的马铃薯根系抗旱相关共表达模块鉴定和核心基因发掘. 作物学报, 2020, 46(7): 1033-1051.
doi: 10.3724/SP.J.1006.2020.94130 |
|
|
|
| [23] |
王文娟, 师尚礼, 康文娟, 杜媛媛, 殷辰. 干旱胁迫下陇中紫花苜蓿对外源精胺的生理响应. 中国农业科学, 2025, 58(4): 676-691. doi: 10.3864/j.issn.0578-1752.2025.10.003.
|
|
|
|
| [24] |
陈彩锦, 马琳, 包明芳, 张国辉, 蒋庆雪, 杨天辉, 王川, 王晓春, 高婷, 王学敏, 刘文辉.111份紫花苜蓿种质资源萌发期抗旱性鉴定评价. 中国农业科学, 2025, 58(10): 1896-1907. doi: 10.3864/j.issn.0578-1752.2025.10.003.
|
|
|
|
| [25] |
张翠梅. 不同抗旱性紫花苜蓿响应干旱的生理及分子机制研究[D]. 兰州: 甘肃农业大学, 2019.
|
|
|
|
| [26] |
夏友霖. 花生晚斑病抗性遗传特性研究[D]. 雅安: 四川农业大学, 2014.
|
|
|
|
| [27] |
doi: 10.1038/nbt.1883 pmid: 21572440 |
| [28] |
doi: 10.1111/jipb.12079 |
| [29] |
|
| [30] |
陈丽琦, 严朋飞, 贾维嘉, 区智, 屈燕. 威氏绿绒蒿(Meconopsis wilsonii)花色相关基因MwF3H的克隆及表达分析. 基因组学与应用生物学, 2022, 41(4): 854-861.
|
|
|
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [1] | 王忠妮, 雷月, 李佳丽, 宫彦龙, 朱速松. ABC转运蛋白OsARG1调控水稻抽穗期的功能[J]. 中国农业科学, 2026, 59(1): 1-16. |
| [2] | 王思琪, 邹利人, 白瑞雯, 闫可, 王思洋, 齐晓光, 申海林, 温景辉. 赤霉素调控‘蜜汁’葡萄穗轴硬化关键基因的挖掘[J]. 中国农业科学, 2026, 59(1): 179-189. |
| [3] | 张义茹, 韩雪, 姚鑫杰, 冯军, 魏爱丽, 李文超, 张彬, 韩渊怀, 李红英. 基于多组学解析谷子后熟米色变化的分子机制[J]. 中国农业科学, 2025, 58(9): 1702-1718. |
| [4] | 潘丽媛, 王永军, 李海军, 侯富, 李菁, 李丽丽, 孙苏阳. 基于转录组和WGCNA筛选小麦籽粒蛋白质累积相关调控基因[J]. 中国农业科学, 2025, 58(6): 1065-1082. |
| [5] | 邹晓威, 夏蕾, 朱晓敏, 孙辉, 周琦, 齐霁, 张亚封, 郑岩, 姜兆远. 基于转录组测序的玉米瘤黑粉菌UM01240过表达菌株诱导玉米抗病性分析[J]. 中国农业科学, 2025, 58(6): 1116-1130. |
| [6] | 孙萍, 朱文灿, 林贤锐, 吴嘉颀, 曹译文, 陈辰斐, 王轶, 朱建锡, 贾惠娟, 钱敏杰, 沈建生. 基于代谢组和转录组解析多雨寡照对桃果皮着色和类黄酮积累的影响[J]. 中国农业科学, 2025, 58(6): 1173-1194. |
| [7] | 谢露露, 李福, 张思远, 高建昌. 基于跨物种转录组解析影响不定根发生的保守基因[J]. 中国农业科学, 2025, 58(6): 1195-1209. |
| [8] | 高岩浩, 王婷婷, 白卫卫, 杜兴杰, 刘贤, 秦本源, 付彤, 孙宇, 高腾云, 张天留. 脂质组与转录组联合揭示南阳牛不同肌肉组织脂质特征的差异表达模式[J]. 中国农业科学, 2025, 58(6): 1239-1258. |
| [9] | 王文娟, 师尚礼, 康文娟, 杜媛媛, 殷辰. 干旱胁迫下陇中紫花苜蓿对外源精胺的生理响应[J]. 中国农业科学, 2025, 58(4): 676-691. |
| [10] | 王凡, 刘陈玮, 陆红臣, 徐仁超, 卞晓春. 蚕豆响应交链格孢侵染的转录组分析及VfPR4的抗病功能验证[J]. 中国农业科学, 2025, 58(22): 4656-4672. |
| [11] | 高荣, 李恒宇, 陈丽娟, 马晖玲. 5‑AzaC缓解紫花苜蓿盐碱胁迫的生理效应及其对DNA甲基化酶基因表达的影响[J]. 中国农业科学, 2025, 58(21): 4482-4496. |
| [12] | 吕缓缓, 李如月, 刘青松, 许蕾, 徐嫣然, 于浩洁, 郭长虹, 龙瑞才. 紫花苜蓿MsKTI3基因克隆及耐盐功能分析[J]. 中国农业科学, 2025, 58(21): 4497-4511. |
| [13] | 杨永念, 曾祥翠, 刘青松, 李如月, 龙瑞才, 陈林, 王雪, 何飞, 康俊梅, 李明娜. 盐碱胁迫下的紫花苜蓿幼苗蛋白组差异分析[J]. 中国农业科学, 2025, 58(21): 4512-4527. |
| [14] | 黄红梅, 王思琦, 杨青川, 郭长虹, 王雪. 磷转运蛋白MsPT5调控紫花苜蓿磷吸收利用[J]. 中国农业科学, 2025, 58(21): 4544-4556. |
| [15] | 张帆, 杨青川. 紫花苜蓿育种历史、现状与展望[J]. 中国农业科学, 2025, 58(21): 4471-4481. |
|
||