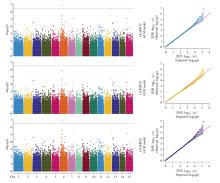
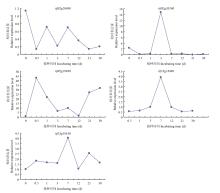
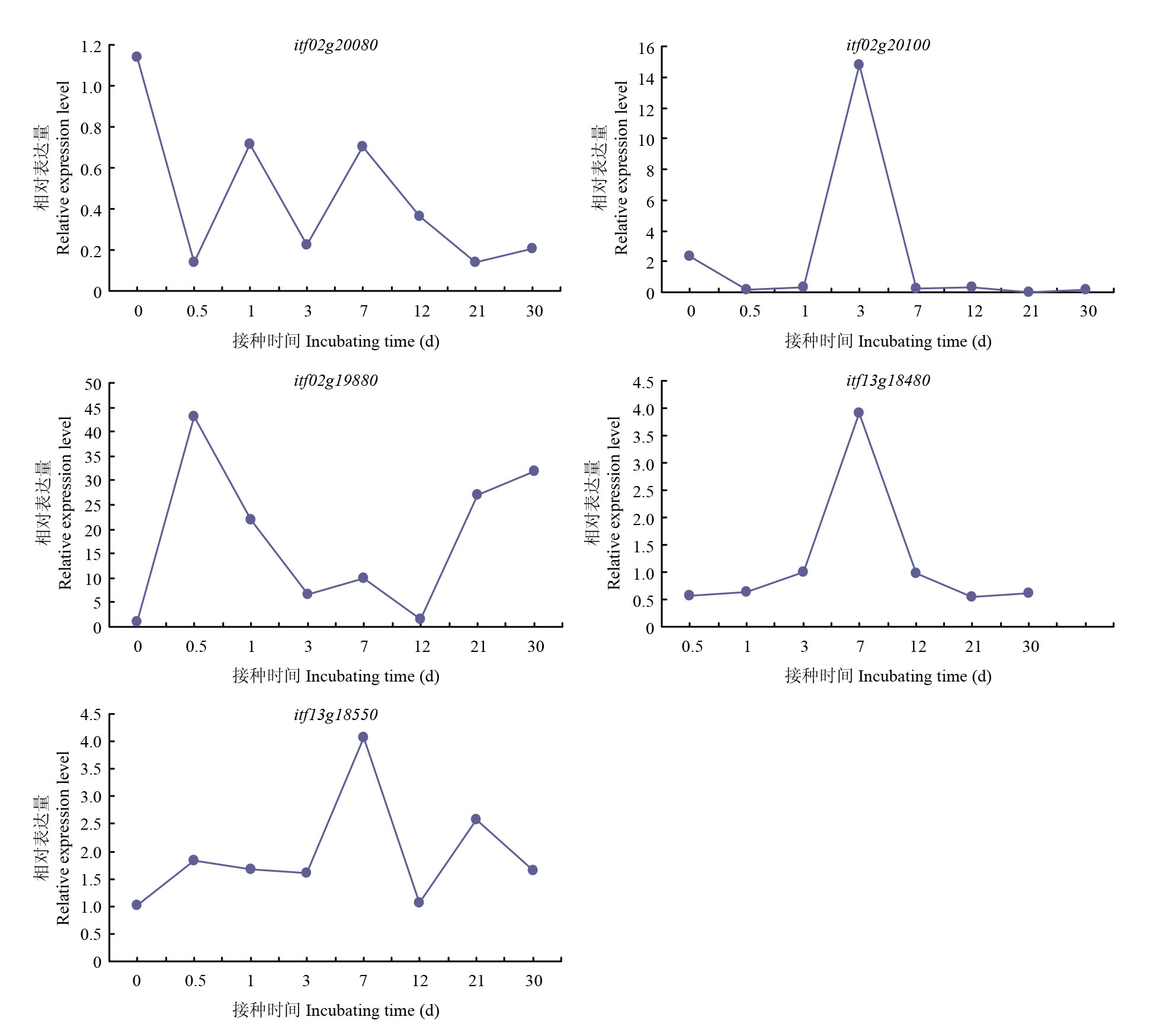
| [1] |
RODRIGUEZ-BONILLA L, CUEVAS H E, MONTERO-ROJAS M, BIRD-PICO F, LUCIANO-ROSARIO D, SIRITUNGA D. Assessment of genetic diversity of sweet potato in Puerto Rico. PLoS ONE, 2014, 9(12): e116184.
|
| [2] |
ZHAO D L, WU S, DAI X B, SU Y J, DAI S B, ZHANG A, ZHOU Z L, TANG J, CAO Q H. QTL analysis of root diameter in a wild diploid relative of sweetpotato (Ipomoea batatas (L.) Lam.) using a SNP-based genetic linkage map generated by genotyping-by- sequencing. Genetic Resources and Crop Evolution, 2021, 68(4): 1375-1388.
|
| [3] |
OKADA Y, KOBAYASHI A, TABUCHI H, KURANOUCHI T. Review of major sweetpotato pests in Japan, with information on resistance breeding programs. Breeding Science, 2017, 67(1): 73-82.
|
| [4] |
马居奎, 张成玲, 杨冬静, 谢逸萍, 孙厚俊. 甘薯腐烂茎线虫重组酶聚合酶扩增方法的建立与应用. 西南农业学报, 2021, 34(2): 293-298.
|
|
MA J K, ZHANG C L, YANG D J, XIE Y P, SUN H J. Development and application of recombinase polymerase amplification assay for rapid detection of Ditylenchus destructor. Southwest China Journal of Agricultural Sciences, 2021, 34(2): 293-298. (in Chinese)
|
| [5] |
赵洪海, 梁晨, 张浴, 段方猛, 宋雯雯, 史倩倩, 黄文坤, 彭德良. 腐烂茎线虫(Ditylenchus destructor Thorne, 1945)生物学研究进展. 生物技术通报, 2021, 37(7): 45-55.
|
|
ZHAO H H, LIANG C, ZHANG Y, DUAN F M, SONG W W, SHI Q Q, HUANG W K, PENG D L. Research advances of biology in Ditylenchus destructor thorne, 1945. Biotechnology Bulletin, 2021, 37(7): 45-55. (in Chinese)
|
| [6] |
姜培, 冯晓东, 王晓亮, 朱莉, 秦萌, 李潇楠, 闫硕. 近年来我国腐烂茎线虫为害与防控形势. 中国植保导刊, 2020, 40(7): 87-90.
|
|
JIANG P, FENG X D, WANG X L, ZHU L, QIN M, LI X N, YAN S. Analysis of the occurrence and control of Ditylenchus destructor in China in recent years. China Plant Protection, 2020, 40(7): 87-90. (in Chinese)
|
| [7] |
刘霞霞, 坚晋卓, 徐鹏刚, 刘永刚, 李惠霞. 甘肃岷县当归茎线虫病发生情况调查分析. 草原与草坪, 2015, 35(4): 1-4.
|
|
LIU X X, JIAN J Z, XU P G, LIU Y G, LI H X. A survey of the Ditylenchus destructor on Angelica in Min County of Gansu Province. Grassland and Turf, 2015, 35(4): 1-4. (in Chinese)
|
| [8] |
马娟, 李秀花, 高波, 王容燕, 陈书龙. 噻唑膦与氨基寡糖素联合应用对甘薯茎线虫病的防治效果. 河北农业科学, 2022, 26(6): 50-55.
|
|
MA J, LI X H, GAO B, WANG R Y, CHEN S L. Control effect of combined application of thiazolidine and aminooligosaccharin on sweetpotato stem nematode disease. Journal of Hebei Agricultural Sciences, 2022, 26(6): 50-55. (in Chinese)
|
| [9] |
马娟, 高波, 李秀花, 王容燕, 黄山, 陈书龙. 噻唑膦与硅肥联合使用对甘薯茎线虫病的防治效果. 植物保护, 2024, 50(2): 307-312, 324.
|
|
MA J, GAO B, LI X H, WANG R Y, HUANG S, CHEN S L. Control efficacy of fosthiazate and silicon fertilizer on Ditylenchus destructor disease. Plant Protection, 2024, 50(2): 307-312, 324. (in Chinese)
|
| [10] |
刘强, 刘环环, 吴春娟, 聂庭彬, 王瑞文. 生物菌剂在甘薯茎线虫病防治中的应用效果. 中国植保导刊, 2023, 43(8): 89-92.
|
|
LIU Q, LIU H H, WU C J, NIE T B, WANG R W. Comparison of effects of different biological agents on the control of sweet potato stem nematode disease. China Plant Protection, 2023, 43(8): 89-92. (in Chinese)
|
| [11] |
QIAO S C, MA J K, WANG Y N, CHEN J W, KANG Z H, BIAN Q Q, CHEN J J, YIN Y M, CAO G Z, ZHAO G R, YANG G H, SUN H J, YANG Y F. Integrated transcriptome and metabolome analyses reveal details of the molecular regulation of resistance to stem nematode in sweet potato. Plants, 2023, 12(10): 2052.
|
| [12] |
闫会, 张成玲, 张允刚, 马居奎, 马猛, 孙厚俊, 李强. 甘薯茎线虫病抗扩展性遗传特性分析与QTL定位. 植物遗传资源学报, 2023, 24(6): 1766-1777.
|
|
YAN H, ZHANG C L, ZHANG Y G, MA J K, MA M, SUN H J, LI Q. Analysis of resistance inheritance and QTL mapping of sweet potato stem nematode disease. Journal of Plant Genetic Resources, 2023, 24(6): 1766-1777. (in Chinese)
|
| [13] |
ZHAO D L, XIAO S Z, ZHANG A, ZHAO L X, DAI X B, YUAN R, WANG J, WANG Y, LI Q L, ZHOU Z L. Construction of high-density genetic map based on SLAF-seq and QTL analysis of major traits in sweetpotato [Ipomoea batatas (L.) Lam. Plant Physiology and Biochemistry, 2024, 211: 108647.
|
| [14] |
WU S, LAU K H, CAO Q H, HAMILTON J P, SUN H H, ZHOU C X, ESERMAN L, GEMENET D C, OLUKOLU B A, WANG H Y, et al. Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nature Communications, 2018, 9: 4580.
|
| [15] |
YIN L L, ZHANG H H, TANG Z S, XU J Y, YIN D, ZHANG Z W, YUAN X H, ZHU M J, ZHAO S H, LI X Y, LIU X L. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics, Proteomics & Bioinformatics, 2021, 19(4): 619-628.
|
| [16] |
吴政, 蒋文翔, 胡霞菲, 张泽霖, 王晓清, 叶晴, 贺浩华, 胡丽芳. ABC转运蛋白对植物花药发育的影响. 分子植物育种, 2024: 1-11. (2024-08-20). https://.cnki.net/kcms/detail/46.1068.S.20240312.1759.022.html.
|
|
WU Z, JIANG W X, HU X F, ZHANG Z L, WANG X Q, YE Q, HE H H, HU L F. Effects of ABC transporter on anther development in plants. Molecular Plant Breeding, 2024: 1-11. (2024-08-20). https://kns.cnki.net/kcms/detail/46.1068.S.20240312.1759.022.html. (in Chinese)
|
| [17] |
张嘉, 李启东, 李翠, 王庆海, 侯新村, 赵春桥, 李树和, 郭强. 植物MATE转运蛋白研究进展. 植物学报, 2023, 58(3): 461-474.
|
|
ZHANG J, LI Q D, LI C, WANG Q H, HOU X C, ZHAO C Q, LI S H, GUO Q. Research progress on MATE transporters in plants. Chinese Bulletin of Botany, 2023, 58(3): 461-474. (in Chinese)
|
| [18] |
MOWBRAY S L, ELFSTRÖM L T, AHLGREN K M, ANDERSSON C E, WIDERSTEN M. X-ray structure of potato epoxide hydrolase sheds light on substrate specificity in plant enzymes. Protein Science, 2006, 15(7): 1628-1637.
|
| [19] |
宋佳乐, 刘洋洋, 司增志, 刘云峰, 吉志新, 乔亚科, 朱英波, 靳恒星. 不同甘薯品种(系)对茎线虫病的抗性鉴定及评价. 江西农业学报, 2023, 35(1): 116-123.
|
|
SONG J L, LIU Y Y, SI Z Z, LIU Y F, JI Z X, QIAO Y K, ZHU Y B, JIN H X. Identification and evaluation of resistance of different sweet potato varieties(species) to Ditylenchus destructor. Acta Agriculturae Jiangxi, 2023, 35(1): 116-123. (in Chinese)
|
| [20] |
李爱贤, 刘庆昌, 王庆美, 翟红, 阎文昭, 张海燕, 李明. 甘薯淀粉含量的QTL定位. 分子植物育种, 2010, 8(3): 516-520.
|
|
LI A X, LIU Q C, WANG Q M, ZHAI H, YAN W Z, ZHANG H Y, LI M. Mapping QTLs for starch content in sweetpotato. Molecular Plant Breeding, 2010, 8(3): 516-520. (in Chinese)
|
| [21] |
蒲志刚, 王大一, 谭文芳, 吴洁, 阎文昭. 利用AFLP构建甘薯连锁图及淀粉含量QTL定位. 西南农业学报, 2010, 23(4): 1047-1050.
|
|
PU Z G, WANG D Y, TAN W F, WU J, YAN W Z. AFLP maps and QTL analysis of starch content of sweet potato. Southwest China Journal of Agricultural Sciences, 2010, 23(4): 1047-1050. (in Chinese)
|
| [22] |
|
|
TANG D B, ZHANG K, LÜ C W, XIE D B, FU T H, WANG J C. Genetic linkage map construction based on EST-SSR and analysis of QTLs for starch content in sweetpotato ( Ipomoea batatas (L.) Lam. Scientia Agricultura Sinica, 2016, 49(23): 4488-4506. doi: 10.3864/j.issn.0578-1752.2016.23.003. (in Chinese)
|
| [23] |
吴洁, 谭文芳, 何俊蓉, 蒲志刚, 王大一, 张正圣, 詹付凤, 阎文昭. 甘薯SRAP连锁图构建淀粉含量QTL检测. 分子植物育种, 2005, 3(6): 841-845.
|
|
WU J, TAN W F, HE J R, PU Z G, WANG D Y, ZHANG Z S, ZHAN F F, YAN W Z. Construct on of SRAP linkage map and QTL mapping for starch content in sweet potato. Molecular Plant Breeding, 2005, 3(6): 841-845. (in Chinese)
|
| [24] |
ZHAO N, YU X X, JIE Q, LI H, LI H, HU J, ZHAI H, HE S Z, LIU Q C. A genetic linkage map based on AFLP and SSR markers and mapping of QTL for dry-matter content in sweetpotato. Molecular Breeding, 2013, 32(4): 807-820.
|
| [25] |
李爱贤, 申春云, 王庆美, 解备涛, 侯夫云, 董顺旭, 张海燕, 张立明. 甘薯β-胡萝卜素含量的QTL定位. 分子植物育种, 2014, 12(2): 270-277.
|
|
LI A X, SHEN C Y, WANG Q M, XIE B T, HOU F Y, DONG S X, ZHANG H Y, ZHANG L M. Mapping QTLs for carotene content in sweetpotato (Ipomoea batatas (L.) Lam.). Molecular Plant Breeding, 2014, 12(2): 270-277. (in Chinese)
|
| [26] |
CERVANTES-FLORES J C, SOSINSKI B, PECOTA K V, MWANGA R O M, CATIGNANI G L, TRUONG V D, WATKINS R H, ULMER M R, YENCHO G C. Identification of quantitative trait loci for dry-matter, starch, and β-carotene content in sweetpotato. Molecular Breeding, 2011, 28(2): 201-216.
|
| [27] |
LI H, ZHAO N, YU X X, LIU Y X, ZHAI H, HE S Z, LI Q, MA D F, LIU Q C. Identification of QTLs for storage root yield in sweetpotato. Scientia Horticulturae, 2014, 170: 182-188.
|
| [28] |
CHANG K Y, LO H, LAI Y, YAO P, LIN K, HWANG S. Identification of quantitative trait loci associated with yield-related traits in sweet potato (Ipomoea batatas). Botanical Studies, 2009, 50(1): 43-55.
|
| [29] |
SHIRASAWA K, TANAKA M, TAKAHATA Y, MA D F, CAO Q H, LIU Q C, ZHAI H, KWAK S S, JEONG J C, YOON U H, LEE H U, HIRAKAWA H, ISOBE S. A high-density SNP genetic map consisting of a complete set of homologous groups in autohexaploid sweetpotato (Ipomoea batatas). Scientific Reports, 2017, 7: 44207.
|
| [30] |
YAN H, MA M, AHMAD M Q, ARISHA M H, TANG W, LI C, ZHANG Y G, KOU M, WANG X, GAO R F, SONG W H, LI Z Y, LI Q. High-density single nucleotide polymorphisms genetic map construction and quantitative trait locus mapping of color-related traits of purple sweet potato [Ipomoea batatas (L.) Lam. Frontiers in Plant Science, 2022, 12: 797041.
|
| [31] |
SASAI R M, TABUCHI H, SHIRASAWA K, KISHIMOTO K, SATO S, OKADA Y, KURAMOTO A, KOBAYASHI A, ISOBE S, TAHARA M, MONDEN Y. Development of molecular markers associated with resistance to Meloidogyne incognita by performing quantitative trait locus analysis and genome-wide association study in sweetpotato. DNA Research, 2019, 26(5): 399-409.
|
| [32] |
MA Z M, GAO W C, LIU L F, LIU M H, ZHAO N, HAN M K, WANG Z, JIAO W J, GAO Z Y, HU Y Y, LIU Q C. Identification of QTL for resistance to root rot in sweetpotato (Ipomoea batatas (L.) Lam) with SSR linkage maps. BMC Genomics, 2020, 21(1): 366.
|
| [33] |
KRETZSCHMAR T, BURLA B, LEE Y, MARTINOIA E, NAGY R. Functions of ABC transporters in plants. Essays in Biochemistry, 2011, 50(1): 145-160.
|
| [34] |
ZHANG C Y, FANG H, WANG J S, TAO H, WANG D B, QIN M C, HE F, WANG R Y, WANG G L, NING Y S. The rice E3 ubiquitin ligase-transcription factor module targets two trypsin inhibitors to enhance broad-spectrum disease resistance. Developmental Cell, 2024, 59(15): 2017-2033.
|







 ), 马居奎, 肖世卓, 周志林, 赵凌霄, 王洁, 戴习彬, 孙厚俊, 曹清河
), 马居奎, 肖世卓, 周志林, 赵凌霄, 王洁, 戴习彬, 孙厚俊, 曹清河