






中国农业科学 ›› 2020, Vol. 53 ›› Issue (16): 3333-3343.doi: 10.3864/j.issn.0578-1752.2020.16.011
高源( ),王大江,王昆(),丛佩华(),李连文,朴继成
),王大江,王昆(),丛佩华(),李连文,朴继成
收稿日期:2019-12-30
接受日期:2020-06-10
出版日期:2020-08-16
发布日期:2020-08-27
通讯作者:
王昆,丛佩华
作者简介:高源,E-mail:基金资助:
GAO Yuan(),WANG DaJiang,WANG Kun(),CONG PeiHua(),LI LianWen,PIAO JiCheng
Received:2019-12-30
Accepted:2020-06-10
Online:2020-08-16
Published:2020-08-27
Contact:
Kun WANG,PeiHua CONG
摘要:
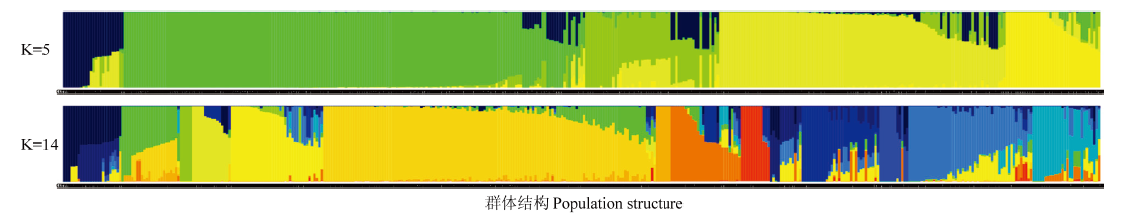
【目的】基于高通量简化基因组测序技术在全基因组开发的SNP标记,对中国原产苹果属植物种内和种间的亲缘关系和群体遗传结构解析,为苹果属植物的起源演化以及系统分类提供理论依据。【方法】对427份15种中国原产苹果属植物种质资源进行高通量简化基因组测序(SLAF-seq),基于获得的SLAF标签,利用BWA软件将其通过比对定位到参考基因组上并获得多态性SLAF标签;利用GATK和SAMtools两种方法在多态性SLAF中开发多态性单核苷酸(SNP),筛选两种方法共同得到的SNP作为开发的SNP标记数据集。根据完整度>0.94、次要等位基因频率(MAF)>0.05过滤筛选获得多态性的SNP。基于筛选多态性SNP,使用MEGA7的NJ(neighbor-joining)算法,构建苹果属不同种的系统进化树。利用Admixture软件进行群体遗传结构分析,假设样品的分群数(K)为1—15进行聚类,根据交叉验证错误率确定最佳K值,解析苹果属不同种间和种内的遗传结构。【结果】通过SLAF-seq技术对427份苹果属植物种质进行测序,最终获得586 454个SLAF标签,其中多态性SLAF标签463 612个。经过序列比对分析和筛选得到46 460个SNP位点,基于这些SNP位点构建苹果属植物不同种的系统发生树并分析群体结构。系统发育分析将15种苹果属植物分成4个类群,群体遗传结构在K=5和K=14为两个分群关键点。综合两种分析方法的结果,15种苹果属植物可分为4个基本的类群,分别为山荆子类群,新疆野苹果和少数中国苹果类群,变叶海棠、花叶海棠、陇东海棠、山楂海棠、滇池海棠和沧江海棠类群,4个苹果属植物栽培种中国苹果、八棱海棠、花红和楸子类群。中国苹果的部分种质中有新疆野苹果和山荆子的基因背景,但其中还有一部分种质可以独立代表类群基因库,其基因库中并没有新疆野苹果的参与,而与山荆子、花红、楸子和八棱海棠密切相关。【结论】利用SLAF技术快速发掘覆盖全基因组的46 460个多态性SNP标记可有效地对中国原产苹果属植物种内和种间的亲缘关系及遗传结构进行研究,为苹果属植物种质资源的鉴定评价、遗传多样性、系统分类和起源演化提供参考。15种苹果属植物分为4个基本类群,苹果属植物野生种和栽培种分类群明显,中国苹果与其他栽培种的亲缘关系密切。
高源,王大江,王昆,丛佩华,李连文,朴继成. 基于高密度SNP标记的苹果属15种植物资源的亲缘关系与遗传结构分析[J]. 中国农业科学, 2020, 53(16): 3333-3343.
GAO Yuan,WANG DaJiang,WANG Kun,CONG PeiHua,LI LianWen,PIAO JiCheng. Genetic Relationship and Structure Analysis of 15 Species of Malus Mill. Based on SNP Markers[J]. Scientia Agricultura Sinica, 2020, 53(16): 3333-3343.
表1
用于SLAF测序分析的15个种苹果属植物种质资源"
| 序号 Code | 供试种 Species | 来源地 Origin | 数量 Number | 序号 Code | 供试种 Species | 来源地 Origin | 数量 Number | |
|---|---|---|---|---|---|---|---|---|
| 1 | 新疆野苹果 Malus sieversii | 新疆 Xinjiang | 161 | 5 | 花红 Malus asiatica | 黑龙江 Heilongjiang | 7 | |
| 2 | 中国苹果 Malus domestica subsp.chinensis | 新疆 Xinjiang | 2 | 甘肃 Gansu | 1 | |||
| 黑龙江 Heilongjiang | 2 | 河北 Hebei | 9 | |||||
| 甘肃 Gansu | 4 | 云南 Yunnan | 1 | |||||
| 河北 Hebei | 14 | 6 | 八棱海棠 Malus robusta | 河北 Hebei | 32 | |||
| 山西 Shanxi | 10 | 山西 Shanxi | 1 | |||||
| 山东 Shandong | 1 | 吉林 Jilin | 1 | |||||
| 3 | 山荆子 Malus baccata | 黑龙江 Heilongjiang | 47 | 7 | 陇东海棠 Malus kansuensis | 甘肃 Gansu | 4 | |
| 甘肃 Gansu | 3 | 8 | 垂丝海棠 Malus halliana | 甘肃 Gansu | 9 | |||
| 河北 Hebei | 10 | 9 | 山楂海棠 Malus komarovii | 吉林 Jilin | 1 | |||
| 山西 Shanxi | 14 | 10 | 变叶海棠 Malus toringoides | 四川 Sichuan | 2 | |||
| 内蒙古 Inner Mongolia | 41 | 云南 Yunnan | 1 | |||||
| 吉林 Jilin | 19 | 11 | 花叶海棠 Malus transitoria | 四川 Sichuan | 1 | |||
| 4 | 楸子 Malus prunifolia | 黑龙江 Heilongjiang | 5 | 12 | 丽江山荆子 Malus rockii | 云南 Yunnan | 1 | |
| 甘肃 Gansu | 4 | 13 | 滇池海棠 Malus yunnanensis | 云南 Yunnan | 1 | |||
| 河北 Hebei | 2 | 14 | 湖北海棠 Malus hupehensis | 云南 Yunnan | 1 | |||
| 山西 Shanxi | 8 | 15 | 沧江海棠 Malus ombrophila | 云南 Yunnan | 1 | |||
| 内蒙古 Inner Mongolia | 1 | |||||||
| 吉林 Jilin | 5 |
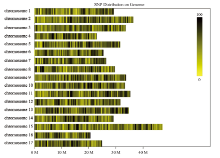
图1
多态性的SNP在染色体上的分布 每一个黄色条带代表一条染色体,黑线代表多态性SNP定位的位置;横坐标为染色体长度,按照1 M的大小对基因组进行划分。黑色越深代表SNP标记数越多,颜色越深的区域即SNP标记集中分布的区域"

表2
基于SNP的苹果属植物15个种间的遗传距离"
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.000 | ||||||||||||||
| 2 | 0.030 | 0.000 | |||||||||||||
| 3 | 0.970 | 1.000 | 0.000 | ||||||||||||
| 4 | 0.600 | 0.606 | 0.394 | 0.000 | |||||||||||
| 5 | 0.030 | 0.000 | 1.000 | 0.606 | 0.000 | ||||||||||
| 6 | 0.030 | 0.000 | 1.000 | 0.606 | 0.000 | 0.000 | |||||||||
| 7 | 0.030 | 0.000 | 1.000 | 0.606 | 0.000 | 0.000 | 0.000 | ||||||||
| 8 | 0.187 | 0.167 | 0.833 | 0.571 | 0.167 | 0.167 | 0.167 | 0.000 | |||||||
| 9 | 0.030 | 0.000 | 1.000 | 0.606 | 0.000 | 0.000 | 0.000 | 0.167 | 0.000 | ||||||
| 10 | 0.953 | 0.981 | 0.019 | 0.398 | 0.981 | 0.981 | 0.981 | 0.821 | 0.981 | 0.000 | |||||
| 11 | 0.143 | 0.120 | 0.880 | 0.581 | 0.120 | 0.120 | 0.120 | 0.247 | 0.120 | 0.866 | 0.000 | ||||
| 12 | 0.030 | 0.000 | 1.000 | 0.606 | 0.000 | 0.000 | 0.000 | 0.167 | 0.000 | 0.981 | 0.120 | 0.000 | |||
| 13 | 0.134 | 0.111 | 0.889 | 0.582 | 0.111 | 0.111 | 0.111 | 0.241 | 0.111 | 0.874 | 0.204 | 0.111 | 0.000 | ||
| 14 | 0.279 | 0.265 | 0.735 | 0.550 | 0.265 | 0.265 | 0.265 | 0.343 | 0.265 | 0.727 | 0.321 | 0.265 | 0.317 | 0.000 | |
| 15 | 0.030 | 0.000 | 1.000 | 0.606 | 0.000 | 0.000 | 0.000 | 0.167 | 0.000 | 0.981 | 0.120 | 0.000 | 0.111 | 0.265 | 0.000 |
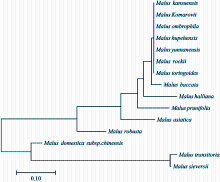
图2
基于SNP位点的苹果属15个种的进化树"
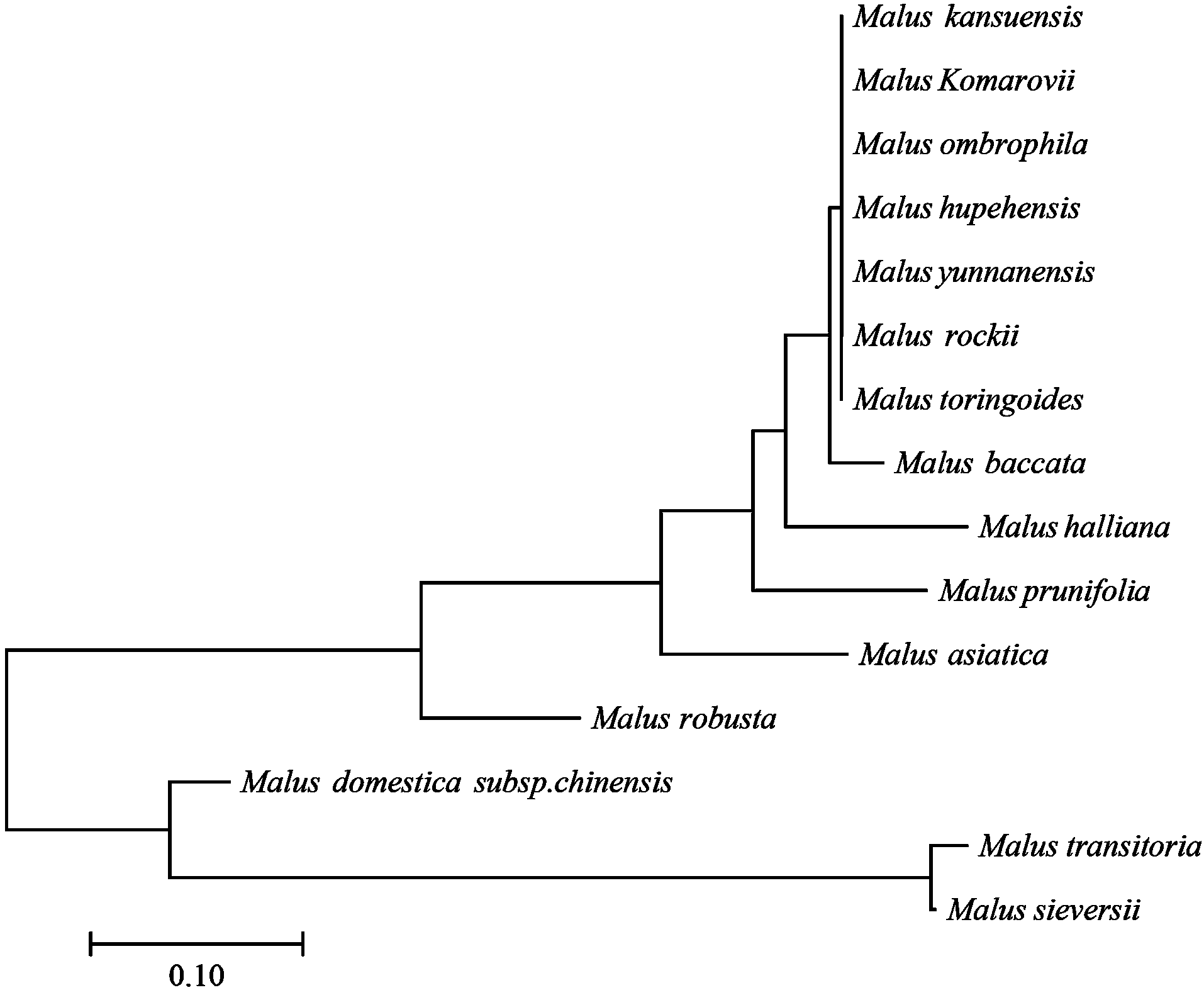
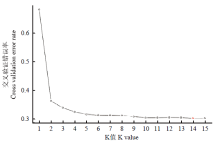
图3
每个K值对应的交叉验证错误率"
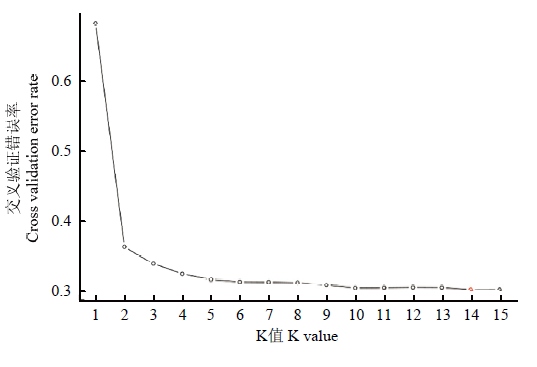

图4
427份15种苹果属植物种质群体遗传结构(K=5和K=14) 每个条柱代表1份种质,横坐标为每个条柱对应的种质编号;一种颜色代表一个类群,纵坐标为Q值0.00—1.00"
| [1] | 俞德俊. 中国果树分类学. 北京: 中国农业出版社, 1979: 91. |
| YU D J. Taxonomy of China Fruits. Beijing: China Agriculture Press, 1979: 91. (in Chinese) | |
| [2] | 贾敬贤, 贾定贤, 任庆棉. 中国作物及其野生近缘植物. 北京: 中国农业出版社, 2006: 40-85. |
| JIA J X, JIA D X, REN Q M. Chinese Crops and Their Wild Relatives. Beijing: China Agriculture Press, 2006: 40-85.(in Chinese) | |
| [3] | 李育农. 苹果属植物种质资源研究. 北京: 中国农业出版社, 2001: 20-72. |
| LI Y N. Researches of Germplasm Resources of Malus Mill. Beijing: China Agricultural Press, 2001: 20-72. (in Chinese) | |
| [4] | BYRNE M, MACDONALD B. Phylogeography and conservation of three oil mallee taxa,Eucalyptus kochii ssp. kochii, ssp.plenissima and E. horistes. Australian Journal of Botany, 2000,48(3):305-312. |
| [5] | 陈国庆, 黄红文, 葛学军. 濒危植物矮沙冬青的等位酶多样性及居群分化. 武汉植物学研究, 2005,23(2):131-137. |
| CHEN G Q, HUANG H W, GE X J. Allozyme diversity and population differentiation in an endangered plant,Ammopiptanthus nanus(Leguminosae). Journal of Wuhan Botanical Research, 2005,23(2):131-137. (in Chinese) | |
| [6] | 潘丽芹, 季华, 陈龙清. 荷叶铁线蕨自然居群的遗传多样性研究. 生物多样性, 2005,13(2):122-129. |
| PAN L Q, JI H, CHEN L Q. Genetic diversity of the natural populations ofAdiantum reniforme var. sinense. Biodiversity Science, 2005,13(2):122-129. (in Chinese) | |
| [7] | BELLUSCI F, PALERMO A M, PELLEGRINO G, MUSACCHIO A. Genetic diversity and spatial structure in the rare, endemic orophyte campanula pseudostenocodon Lac.(Apennines, Italy), as a infered from nuclear and plastid variation. Plant Biosystems, 2008,142(1):24-29. |
| [8] | SHAH A, LI D Z, GAO L M, LI H T, MOLLER M. Genetic diversity within and among populations of the endangered species Taxus fauna(Taxaceae) from Pakistan and implications for its conservation. Biochemical Systematics and Ecology, 2008,36(3):183-193. |
| [9] | 寇淑君, 霍阿红, 付国庆, 纪军建, 王瑶, 左振兴, 刘敏轩, 陆平. 利用荧光SSR分析中国糜子的遗传多样性和群体遗传结构. 中国农业科学, 2019,52(9):1475-1487. |
| KOU S J, HUO A H, FU G Q, JI J J, WANG Y, ZUO Z X, LIU M X, LU P. Genetic diversity and population structure of broomcorn millet in China based on fluorescently labeled SSR. Scientia Agricultura Sinica, 2019,52(9):1475-1487. (in Chinese) | |
| [10] | 张冰冰. 抗寒苹果、梨种质资源遗传多样性研究[D]. 长春: 吉林农业大学, 2008: 1-3. |
| ZHANG B B. Study on the genetic diversity of winterhardy apple and pear germplasm resources[D]. Changchun: Jinlin Agricultural University, 2008: 1-3. (in Chinese) | |
| [11] | 王涛, 祝军, 李光晨, 周爱琴, 张文. 苹果砧木亲缘关系AFLP分析. 中国农业科学, 2002,34(3):256-259. |
| WANG T, ZHU J, LI G C, ZHOU A Q, ZHANG W. AFLP analysis of genetic relationships in apple rootstocks. Scientia Agricultura Sinica, 2002,34(3):256-259. (in Chinese) | |
| [12] | ZHANG D P, CARBAJULCA D, OJEDA L. Microsatellite analysis of genetic diversity in sweet potato varieties from Latin America and the Pacific region: Its implications on the dispersal of the crop. Genetic Resources and Crop Evolution, 2004,51(2):115-120. |
| [13] | 高源, 王昆, 王大江, 赵继荣, 张彩霞, 丛佩华, 刘立军, 李连文, 朴继成. 7个来源地区山荆子的遗传多样性与群体结构分析. 中国农业科学 2018,51(19):3766-3777. |
| GAO Y, WANG K, WANG D J, ZHAO J R, ZHANG C X, CONG P H, LIU L J, LI L W, PIAO J C. The genetic diversity and population structure analysis on Malus baccata(L.) Borkh from 7 Sources. Scientia Agricultura Sinica, 2018,51(19):3766-3777. (in Chinese) | |
| [14] | RAFALSKI A. Applications of single nucleotide polymorphisms in crop genetics. Genome Studies and Molecular Genetics, 2002,5:94-100. |
| [15] |
GUPTA P K, RUSTGI S, MIR R R. Array-based high-throughput DNA markers for crop improvement. Heredity, 2008,101:5-18.
doi: 10.1038/hdy.2008.35 pmid: 18461083 |
| [16] |
CHAGNE D, GASIC K, CROWHURST R N, HAN Y P, BASSETT H C, BOWATTE D R, LAWRENCE T J, RIKKERINK E H A, GARDINER S E, KORBAN S S. Development of a set of SNP markers present in expressed genes of the apple. Genomics, 2008,92(5):353-358.
pmid: 18721872 |
| [17] |
MICHELETTI D, TROGGIO M, ZHARKIKH A, COSTA F, MALNOY M, VELASCO R, SALVI S. Genetic diversity of the genusMalus and implications for linkage mapping with SNPs. Tree Genetics & Genomes, 2011,7:857-868.
doi: 10.1007/s11295-011-0380-8 |
| [18] | 常源升, 孙瑞, 陈东玫, 王忆, 杨凤秋, 赵永波, 张新忠, 韩振海. 苹果果形相关基因的主基因分析与QTL定位. 园艺学报, 2013,40(S):2578. |
| CHANG Y S, SUN R, CHEN D M, WANG Y, YANG F Q, ZHAO Y B, ZHANG X Z, HAN Z H. Major gene analysis and QTL mapping of apple fruit shape related genes. Acta Horticulturae Sinica, 2013,40(S):2578. (in Chinese) | |
| [19] |
SUN R, CHANG Y S, YANG F Q, WANG Y, LI H, ZHAO Y B, CHEN D M, WU T, ZHANG X Z, HAN Z H. A dense SNP genetic map constructed using restriction site-associated DNA sequencing enables detection of QTLs controlling apple fruit quality. BMC Genomics, 2015,16:747.
pmid: 26437648 |
| [20] | 孙瑞. 苹果高密度遗传连锁图谱构建与重要果实品质性状QTL定位[D]. 北京: 中国农业大学, 2015: 3-18. |
| SUN R. High-density genetic linkage map construction and QTL identification for important fruit quality traits in apple[D]. Beijing: China Agricultural University, 2015: 3-18. (in Chinese) | |
| [21] | 刘更森. 苹果SSR和SNP标记开发及在遗传图谱构建和品种鉴定中的应用[D]. 长沙: 湖南农业大学, 2018: 10-15. |
| LIU G S. Development of apple SSR and SNP markers and application in genetic map construction and cultivar identification[D]. Changsha: Hunan Agricultural University, 2018: 10-15. (in Chinese) | |
| [22] | VELASO R, ZHARKIKH A, AFFOURTIT J, DHINGRA A, CESTARO A, KALYANARAMAN A, FONTANA P, BHATNAGAR S K, TROGGIO M, PRUSS D, SALVI S, PINDO M, BALDI P, CASTELLETTI S, CAVAIUOLO M, COPPOLA G, COSTA F, COVA V, DAL R A, GOREMYKIN V, et al. The genome of the domesticated apple(Malus × domestica Borkh.). Nature Genetics, 2010,4:833-839. |
| [23] |
DACCORD N, CELTON J M, LINSMITH G, BECKER C, CHOISNE N, SCHIJLEN E, GEEST H, BIANCO L, MICHELETTI D, VELASCO R, PIERRO E A, GOUZY J, REES D J G, GUERIF P, MURANTY H, DUREL C E, LAURENS F, LESPINASSE Y, GAILLARD S, AUBOURG S, QUESNEVILLE H, WEIGEL D, WEG E, BUCHER E. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nature Genetics, 2017,49(7):1099-1106.
pmid: 28581499 |
| [24] | ZHANG L Y, JIANG H, HAN X L, LI J J, GAO Y, RICHARDS C M, ZHANG C X, TIAN Y, LIU G M, GUL H, WANG D J, TIAN Y, YANG C X, MENG M H, YUAN G P, KANG G D, WU Y L, WANG K, ZHANG H T, WANG D P, CONG P H. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nature Communication, 2019. https://doi.org/ 10.1038/ s41467-019-09518-x.. |
| [25] | 周蓓蓓, 朱海军, 生静雅, 刘广勤. DNA分子标记在果树种质资源遗传多样性研究中的应用. 江西农业学报, 2011,23(9):47-50. |
| ZHOU B B, ZHU H J, SHENG J Y, LIU G Q. Application of DNA molecular markers in genetic diversity study of fruit tree germplasm resources. Acta Agriculturae Jiangxi, 2011,23(9):47-50. (in Chinese) | |
| [26] | 彭强, 叶生鑫, 黄龙, 张大双, 刘颖, 吴健强, 张玉珊, 朱速松. 运用SLAF-seq技术构建水稻高密度遗传图谱. 分子植物育种, 2016,14(8):2127-2132. |
| PENG Q, YE S X, HUANG L, ZHANG D S, LIU Y, WU J Q, ZHANG Y S, ZHU S S. Construction of a high-density genetic map in rice by using specific-length amplified fragment sequencing(SLAF- seq)technology. Molecular Plant Breeding, 2016,14(8):2127-2132. (in Chinese) | |
| [27] | 苏文瑾, 赵宁, 雷剑, 王连军, 柴沙沙, 杨新笋. 基于SLAF-seq 技术的甘薯SNP位点开发. 中国农业科学, 2016,49(1):27-47. |
| SU W J, ZHAO N, LEI J, WANG L J, CHAI S S, YANG X S. SNP sites developed by specific length amplification fragment sequencing(SLAF-seq)in sweetpotato. Scientia Agricultura Sinica, 2016,49(1):27-47. (in Chinese) | |
| [28] | 刘凯, 李开祥, 韦晓娟, 梁文汇, 王坤. 基于SLAF-seq技术的金花茶SNP 标记开发及遗传分析. 经济林研究, 2019,3(3):79-83. |
| LIU K, LI K X, WEI X J, LIANG W H, WANG K. Development and genetic analysis on SNP sites from Camellia nitidssima based on SLAF-seq technology. Non-wood Forest Research, 2019,3(3):79-83. (in Chinese) | |
| [29] | 张恒, 刘众杰, 樊秀彩, 张川, 崔力文, 刘崇怀, 房经贵. 葡萄果粒形状简化基因组关联分析. 园艺学报, 2017,44(10):1959-1968. |
| ZHANG H, LIU Z J, FAN X C, ZHANG C, CUI L W, LIU C H, FANG J G. Genome-wide association mapping of berry shape traits via the reduced representation sequencing in grape. Acta Horticulturae Sinica, 2017,44(10):1959-1968. (in Chinese) | |
| [30] | 陶红霞. 基于SLAF标记的苹果遗传连锁图谱构建[D]. 杨凌: 西北农林科技大学, 2015: 9-12. |
| TAO H X. The genetic linkage map construction of apple based on SLAF marker [D]. Yangling: Northwest A & F University, 2015: 9-12. (in Chinese) | |
| [31] |
LI H, DURBIN R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009,25(14):1754-1760.
pmid: 19451168 |
| [32] |
MCKENNA A, HANNA M, BANKS E, SIVACHENKO A, CIBULSKIS K, KERNYTAKY A, GARIMELLA K, ALTSHULER D, GABRIEL S, DALY M, DEPRISTO M A. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 2010,20(9):1297-1303.
pmid: 20644199 |
| [33] |
LI H, HANDSAKER B, WYSOKER A, FENNELL T, RUAN J, HOMER N, DURBIN R. 1000 GENOME PROJECT DATA PROCESSING SUBGROUP. The sequence alignment/map(SAM)format and SAMtools. Bioinformatics, 2009,25(16):2078-2079.
doi: 10.1093/bioinformatics/btp352 pmid: 19505943 |
| [34] | SUNSERI F, LUPINI A, MAUCERI A, DE LORENZIS G, ARANITI F, BRANCADORO L, MERCATI F. Single nucleotide polymorphism profiles reveal an admixture genetic structure of grapevine germplasm from Calabria, Italy, uncovering its key role for the diversification of cultivars in the Mediterranean Basin. Australian Journal of Grape and Wine Research, 2018,24(3):345-359. |
| [35] | KUMAR S, STECHER G, TAMURA K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Molecular Biology and Evolution, 2016,33(7):1870-1874. |
| [36] |
SAITOU N, NEI M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 1987,4(4):406-425.
pmid: 3447015 |
| [37] |
ALEXANDER D H, NOVEMBRE J, LANGE K. Fast model-based estimation of ancestry in unrelated individuals. Genome Research, 2009,19(9):1655-1664.
pmid: 19648217 |
| [38] | 邹喻苹, 葛颂. 新一代分子标记-SNPs及其应用. 生物多样性, 2003,11(5):370-382. |
| ZOU Y P, GE S. A novel molecular marker-SNPs and its application. Biodiversity Science, 2003,11(5):370-382. (in Chinese) | |
| [39] |
WANG D G, FAN J B, SIAO C J, BERNO A, YOUNG P, SAPOLSKY R, GHANDOUR G, PERKINS N, WINCHESTER E, SPENCER J, KRUGLYAK L, STEIN L, HSIE L, TOPALOGLOU T, HUBBELL E, ROBINSON E, MITTMANN M, MORRIS M S, SHEN N P, KILBURN D, RIOUX J, NUSBAUM C, ROZEN S, HUDSON T J, LIPSHUTZ R, CHEE M, LANDER E S. Large-Scale identification, mapping and genotyping of single-nucleotide polymorphisms in the human genome. Science, 1998,280(5366):1077-1082.
pmid: 9582121 |
| [40] |
SYYNEN A C. Accessing genetic variation: Genotyping single nucleotide polymorphisms. Nature Reviews Genetics, 2001,2:930-942.
pmid: 11733746 |
| [41] |
WILTSHIRE T, PLETCHER W T, BATALOV S, BARNES S W, TARANTINO L M, COOKE M P, WU H, SMYLIE K, SANTROSYAN A, COPELAND N G, JENKINS N A, KALUSH F, MURAL R J, GLYNNE R J, KAY S A, ADAMS M D, FLETCHER C F. Genome- wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proceeding of the National Academy of Sciences, 2003,100(6):3380-3385.
doi: 10.1073/pnas.0130101100 |
| [42] |
JANDER G, NORRIS S R, ROUNSLEY S D, BUSH D F, LEVIN I M, LAST R L. Arabidopsis map-based cloning in the post-genome era. Plant Physiology, 2002,129:440-450.
pmid: 12068090 |
| [43] |
FELTUIS F A, WAN J, SCHULZE S R, ESTILL J C, JIANG N, PATERSONET A H. An SNP resources for rice genetics and breeding based on subspecies Indica and Japonica genome alignments. Genome Research, 2004,14(9):1812-1819.
doi: 10.1101/gr.2479404 pmid: 15342564 |
| [44] |
SOMERS D J, KIRKPATRICK R, MONIWA M, WALSH A. Mining single-nucleotide polymorphisms from hexaploid wheat ESTs. Genome, 2003,46(3):431-437.
doi: 10.1139/g03-027 pmid: 12834059 |
| [45] |
赵久然, 李春辉, 宋伟, 王元东, 张如养, 王继东, 王凤格, 田红丽, 王蕊. 基于SNP芯片揭示中国玉米育种种质的遗传多样性与群体遗传结构. 中国农业科学, 2018,51(4):626-634.
doi: 10.3864/j.issn.0578-1752.2018.04.003 |
|
ZHAO J R, LI C H, SONG W, WANG Y D, ZHANG R Y, WANG J D, WANG F G, TIAN H L, WANG R. Genetic diversity and population structure of important Chinese maize breeding germplasm revealed by SNP-Chips. Scientia Agricultura Sinica, 2018,51(4):626-634. (in Chinese)
doi: 10.3864/j.issn.0578-1752.2018.04.003 |
|
| [46] |
陈士强, 秦树文, 黄泽峰, 戴毅, 张璐璐, 高营营, 陈建民. 基于SLAF-seq技术开发长穗偃麦草染色体特异分子标记. 作物学报, 2013,39(4):727-734.
doi: 10.3724/SP.J.1006.2013.00727 |
|
CHEN S Q, QIN S W, HUANG Z F, DAI Y, ZHANG L L, GAO Y Y, CHEN J M. Development of specific molecular markers forThinopyrum elongatum chromosome using SLAF-seq technique. Acta Agronomica Sinica, 2013,39(4):727-734. (in Chinese)
doi: 10.3724/SP.J.1006.2013.00727 |
|
| [47] |
CHEN W, YAO J, CHU L, LI Y, GUO X M, ZHANG Y S. The development of specific SNP markers for chromosome 14 in cotton using next-generation sequencing. Plant Breeding, 2014,133(2):256-261.
doi: 10.1111/pbr.12144 |
| [48] |
ZHU Y F, YIN Y F, YANG K Q, LI J H, SANG Y L, HUANG L, FAN S. Construction of a high-density genetic map using specific length amplified fragment markers and identification of a quantitative trait locus for anthracnose in walnut (Juglans regia L.). BMC Genomics, 2015,16(1):1-13.
doi: 10.1186/1471-2164-16-1 |
| [49] |
HAN Y P, ZHAO X, LIU D Y, LI Y H, LIGHTFOOT D A, YANG Z J, ZHAO L, ZHOU G, WANG Z K, HUANG L, ZHANG Z W, QIU L J, ZHENG H K, LI W B. Domestication footprints anchor genomic regions of agronomic importance in soybeans. New Phytologist, 2016,209:871-884.
doi: 10.1111/nph.13626 pmid: 26479264 |
| [50] | 李贝贝, 张恒, 姜建福, 张颖, 樊秀彩, 房经贵, 刘崇怀. 基于SLAF-seq技术的葡萄种质遗传多样性分析. 园艺学报, 2019,46(11):2109-2118. |
| LI B B, ZHANG H, JIANG J F, ZHANG Y, FAN X C, FANG J G, LIU C H. Analysis of genetic diversity of grape germplasms using SLAF-seq technology. Acta Horticulturae Sinica, 2019,46(11):2109-2118. (in Chinese) | |
| [51] |
DUAN N B, BAI Y, SUN H H, WANG N, MA Y M, LI M J, WANG X, JIAO C, LEGALL N, MAO L Y, WAN S B, WANG K, HE T M, FENG S Q, ZHANG Z Y, MAO Z Q, SHEN X, CHEN X L, JIANG Y M, WU S J, YIN C M, GE S F, YANG L, JIANG S H, XU H F, LIU J X, WANG D Y, QU C Z, WANG Y C, ZUO W F, XIANG L, LIU C, ZHANG D Y, GAO Y, XU Y M, XU K N, CHAO T, FAZIO G, SHU H R, ZHONG G Y, CHENG L L, FEI Z J, CHEN X S. Genome re-sequencing reveals the history of apple and supports a two-stage for fruit enlargement. Nature Communication, 2017,8:249.
doi: 10.1038/s41467-017-00336-7 |
| [52] |
CAO K, ZHENG Z J, WANG L R, LIU X, ZHU G R, FANG W C, CHENG S F, ZENG P, CHEN C W, WANG X W, XIE M, ZHONG X, WANG X L, ZHAO P, BIAN C, ZHU Y L, ZHANG J H, MA G S, CHEN C X, LI Y J, HAO F G, LI Y, HUANG G D, LI Y X, LI H Y, XU X, WANG J. Comparative population genomics reveals the domestication history of the peach,Prunus persica, and human influences on perennial fruit crops. Genome Biology, 2014,15:415.
doi: 10.1186/s13059-014-0415-1 pmid: 25079967 |
| [1] | 唐华苹,陈黄鑫,李聪,苟璐璐,谭翠,牟杨,唐力为,兰秀锦,魏育明,马建. 基于55K SNP芯片的普通小麦穗长非条件和条件QTL分析[J]. 中国农业科学, 2022, 55(8): 1492-1502. |
| [2] | 屠云洁,姬改革,章明,刘一帆,巨晓军,单艳菊,邹剑敏,李华,陈智武,束婧婷. 鸡Wnt3a的SNPs筛选及其与皮肤毛囊密度性状关联分析[J]. 中国农业科学, 2022, 55(23): 4769-4780. |
| [3] | 许志英,王佰翠,马晓兰,贾子苗,叶兴国,林志珊,胡汉桥. 基于小麦SNP芯片对簇毛麦6V#2和6V#4染色体及其与小麦6A、6D染色体的多态性分析[J]. 中国农业科学, 2021, 54(8): 1579-1589. |
| [4] | 聂兴华, 郑瑞杰, 赵永廉, 曹庆芹, 秦岭, 邢宇. 利用荧光SSR分子标记评估中国栗属植物遗传多样性[J]. 中国农业科学, 2021, 54(8): 1739-1750. |
| [5] | 樊晓静, 于文涛, 蔡春平, 林浥, 王泽涵, 房婉萍, 张见明, 叶乃兴. 利用SNP标记构建茶树品种资源分子身份证[J]. 中国农业科学, 2021, 54(8): 1751-1760. |
| [6] | 刘有春,刘威生,王兴东,孙斌,刘修丽,杨艳敏,魏鑫,杨玉春,张舵,刘成,李天忠. 基于简化基因组测序的越橘杂交后代鉴定[J]. 中国农业科学, 2021, 54(2): 370-378. |
| [7] | 严勇亮,时晓磊,张金波,耿洪伟,肖菁,路子峰,倪中福,丛花. 春小麦籽粒主要品质性状的全基因组关联分析[J]. 中国农业科学, 2021, 54(19): 4033-4047. |
| [8] | 宋春晖,陈晓菲,王枚阁,郑先波,宋尚伟,焦健,王苗苗,马锋旺,白团辉. 基于SLAF-seq技术鉴定苹果砧木耐涝候选基因[J]. 中国农业科学, 2021, 54(18): 3932-3944. |
| [9] | 王继庆,任毅,时晓磊,王丽丽,张新忠,苏力坛·姑扎丽阿依,谢磊,耿洪伟. 小麦籽粒超氧化物歧化酶(SOD)活性全基因组关联分析[J]. 中国农业科学, 2021, 54(11): 2249-2260. |
| [10] | 张芳,任毅,曹俊梅,李法计,夏先春,耿洪伟. 基于SNP标记的小麦籽粒性状全基因组关联分析[J]. 中国农业科学, 2021, 54(10): 2053-2063. |
| [11] | 盖钧镒,贺建波. 限制性两阶段多位点全基因组关联分析法(RTM-GWAS)的特点、常见提问与应用前景[J]. 中国农业科学, 2020, 53(9): 1699-1703. |
| [12] | 贺建波,刘方东,王吴彬,邢光南,管荣展,盖钧镒. 限制性两阶段多位点全基因组关联分析法在遗传育种中的应用[J]. 中国农业科学, 2020, 53(9): 1704-1716. |
| [13] | 徐云碧,杨泉女,郑洪建,许彦芬,桑志勤,郭子锋,彭海,张丛,蓝昊发,王蕴波,吴坤生,陶家军,张嘉楠. 靶向测序基因型检测(GBTS)技术及其应用[J]. 中国农业科学, 2020, 53(15): 2983-3004. |
| [14] | 何俊,李智,吴晓林. 约束标准化线性回归法估计合成品种动物基因组品种构成[J]. 中国农业科学, 2020, 53(1): 191-200. |
| [15] | 唐裕杰,王刘豪,李凯,李继莲. 我国不同地区熊蜂短膜虫的感染情况及亲缘关系[J]. 中国农业科学, 2019, 52(6): 1110-1118. |
|
||