






中国农业科学 ›› 2020, Vol. 53 ›› Issue (3): 600-611.doi: 10.3864/j.issn.0578-1752.2020.03.012
高源,王大江,王昆( ),丛佩华(),张彩霞,李连文,朴继成
),丛佩华(),张彩霞,李连文,朴继成
收稿日期:2019-07-21
接受日期:2019-11-01
出版日期:2020-02-01
发布日期:2020-02-13
通讯作者:
王昆,丛佩华
作者简介:高源,E-mail:gaoyuan02@caas.cn。
基金资助:
GAO Yuan,WANG DaJiang,WANG Kun(),CONG PeiHua(),ZHANG CaiXia,LI LianWen,PIAO JiCheng
Received:2019-07-21
Accepted:2019-11-01
Online:2020-02-01
Published:2020-02-13
Contact:
Kun WANG,PeiHua CONG
摘要:
【目的】山荆子是中国原产苹果属植物中分布最广泛的种,母系遗传的叶绿体基因组的非编码区适用于较低的分类阶元(如科、属)的系统研究。对野外考察新收集的山荆子种质的叶绿体DNA(cpDNA)非编码区进行测序,解析其序列遗传变异,从母系遗传基因的角度探究山荆子不同居群的遗传多样性和系统演化关系,为我国山荆子种质资源的起源演化以及收集和保护提供理论依据。【方法】利用4对叶绿体DNA引物扩增新收集的215份山荆子种质资源的4个非编码区trnH-psbA、trnS-trnG spacer+intron、trnT-5'trnL和5'trnL-trnF,对每个基因间区正反向测序获得的序列进行人工校对后,使用MEGA 7.0进行序列拼接和比对,并构建山荆子不同居群间基于遗传距离的Neighbour-Joining系统发育树;使用DnaSP ver5.10.01计算叶绿体DNA的遗传多样性参数,计算不同居群间的基因流和基因分化;利用Arlequin v3.5分析标准分子变异(AMOVA);运用NetWork ver4.6.1.2构建种内居群间的叶绿体DNA单倍型邻接网络关联图。【结果】4个叶绿体DNA非编码区经测序、拼接、比对和合并之后的片段长度为3 777 bp,共有171个多态性变异位点,其中包含150个插入-缺失位点、20个简约信息位点和1个单一突变位点。在215份山荆子种质中,trnH-psbA、trnS-trnG spacer + intron、trnT-5'trnL和5'trnL-trnF区域的变异位点数量分别为26、32、103和10个,单倍型数量分别为8、8、6和4个,合并之后的叶绿体DNA片段的单倍型为24个。核苷酸多样性最高的区域为trnT-5'trnL(Pi=0.01174),单倍型(基因)多样性最高的为trnS-trnG spacer+intron(Hd=0.599),最低的为5'trnL-trnF(Hd=0.228)。215份山荆子种质叶绿体DNA多样性较高(Hd=0.727,Pi=0.00577)。Tajima’s D检验中,4个cpDNA区域在各检验水平上均不显著,检测的4个cpDNA区域在进化上遵循中性模型。AMOVA分析表明遗传变异主要存在于群体内部。【结论】供试4个叶绿体DNA非编码区适合苹果属山荆子种质遗传多样性和系统演化分析。在叶绿体DNA水平导致山荆子群体进化的原因不是自然选择,而是突变压力和遗传漂变。群体间遗传分化与其地理距离不完全相关。山荆子可能为多点起源,推测黑龙江和吉林,内蒙古,甘肃和山西为3个可能的起源地区。
高源,王大江,王昆,丛佩华,张彩霞,李连文,朴继成. 基于叶绿体DNA变异的山荆子种质遗传多样性和系统演化[J]. 中国农业科学, 2020, 53(3): 600-611.
GAO Yuan,WANG DaJiang,WANG Kun,CONG PeiHua,ZHANG CaiXia,LI LianWen,PIAO JiCheng. Genetic Diversity and Phylogenetics of Malus baccata (L.) Borkh Revealed by Chloroplast DNA Variation[J]. Scientia Agricultura Sinica, 2020, 53(3): 600-611.
表1
4个叶绿体基因间区及4对叶绿体DNA引物序列"
| 编号 Code | cpDNA间区 cpDNA intergenic regions | F序列(5′-3′) Forward sequence (5′-3′) | R序列(5′-3′) Reverse sequence (5′-3′) | 扩增片段长度 Amplified fragment length |
|---|---|---|---|---|
| CP20 | trnH-psbA | CGCGCATGGTGGATTCACAATCC | GTTATGCATGAACGTAATGCTC | ~400 |
| CP21 | trnS-trnG spacer + intron | AGATAGGGATTCGAACCCTCGGT | GTAGCGGGAATCGAACCCGCATC | ~1500 |
| CP22 | trnT-5' trnL | CATTACAAATGCGATGCTCT | TCTACCGATTTCGCCATATC | ~1200 |
| CP23 | 5' trnL-trnF | ATTTGAACTGGTGACACGAG | CGAAATCGGTAGACGCTACG | ~1000 |

图1
部分苹果样品在叶绿体DNA间区trnH-psbA的PCR扩增产物的2%琼脂糖凝胶电泳图"
表2
山荆子4个叶绿体基因间区的PCR扩增"
| 编号 Code | 扩增区域 Region | PCR反应条件 PCR condition | 退火温度 Annealing temperature |
|---|---|---|---|
| CP20 | trnH-psbA | 80℃ 5 min;94℃ 30 s,56℃ 30 s,72℃ 1 min,35个循环(35 cycles);72℃ 10 min | 56℃ |
| CP21 | trnS-trnG spacer + intron | 80℃ 5 min;96℃ 10 s,50℃ 5 s,60℃ 4 min,30个循环(30 cycles);60℃ 10 min | 50℃ |
| CP22 | trnT-5' trnL | 96℃ 5 min;96℃ 1 min,57℃ 2 min,72℃ 2.5 min,34个循环(34 cycles);72℃ 10 min | 57℃ |
| CP23 | 5' trnL-trnF | 80℃ 5 min;96℃ 10 s,50℃ 5 s,60℃ 4 min,30个循环(30 cycles);60℃ 10 min | 50℃ |
表3
供试山荆子的4个cpDNA区域的多态性信息"
| 叶绿体DNA区域 cpDNA regions | 片段长度 Length of fragment (bp) | 变异位点 Variable sites (Vs) | 单一突变位点 Singleton variable sites (Ss) | 简约信息位点 Parsimony informative sites (Ps) | 插入-缺失位点 Insertion-deletion gaps (Is) | 核苷酸多样性 Nucleotide diversity (Pi) | 平均核苷酸差异 Average number of nucleotide difference (K) |
|---|---|---|---|---|---|---|---|
| trnH-psbA | 285 | 26 | 0 | 2 | 24 | 0.00913 | 3.13236 |
| trnS-trnG spacer+intron | 1383 | 32 | 1 | 5 | 26 | 0.00311 | 4.52649 |
| trnT-5' trnL | 1140 | 103 | 0 | 11 | 92 | 0.01174 | 15.68937 |
| 5' trnL-trnF | 965 | 10 | 0 | 2 | 8 | 0.00045 | 0.44216 |
| 组合 Combined | 3773 | 171 | 1 | 20 | 150 | 0.00577 | 23.79039 |
表4
供试山荆子叶绿体DNA单倍型多样性"
| cpDNA区域 cpDNA Regions | 单倍型数目 Number of haplotypes (h) | 单倍型(基因)多样性 Haplotype (Gene) diversity (Hd) | 单倍型多样性方差 Variance of haplotype Diversity (Vh) | 单倍型多样性标准差 Standard deviation of haplotype diversity (Sh) |
|---|---|---|---|---|
| trnH-psbA | 8 | 0.4762 | 0.00159 | 0.040 |
| trnS-trnG spacer + intron | 8 | 0.599 | 0.00111 | 0.033 |
| trnT-5' trnL | 6 | 0.372 | 0.00168 | 0.041 |
| 5' trnL-trnF | 4 | 0.228 | 0.00133 | 0.037 |
| 组合 Combined | 24 | 0.727 | 0.00084 | 0.029 |
表5
215份山荆子叶绿体 DNA 片段的Tajima’s D值"
| cpDNA区域 cpDNA regions | Tajima's D值 Tajima's D | 显著性 Significance |
|---|---|---|
| trnH-psbA | -0.78313 | P>0.10 |
| trnS-trnG spacer+ intron | -0.45018 | P>0.10 |
| trnT-5' trnL | -0.29083 | P>0.10 |
| 5' trnL-trnF | -1.70516 | 0.05<P<0.10 |
| 组合 Combined | -0.54177 | P>0.10 |
表6
215份山荆子按来源省份的7个群体"
| 试材编号Code of accessions | 来源地Origion | 个体数量Number of accessions | 群体代码Code of populations |
|---|---|---|---|
| MB1-MB83 | 黑龙江 Heilongjiang | 83 | MBHLJ |
| MB84-MB101 | 吉林 Jilin | 18 | MBJL |
| MB102-MB143 | 内蒙古 Inner Mongolia | 42 | MBNM |
| MB144-MB197 | 河北 Hebei | 54 | MBHB |
| MB198-MB210 | 山西 Shanxi | 13 | MBSX |
| MB211-MB214 | 甘肃 Gansu | 4 | MBGS |
| MB215 | 辽宁 Liaoning | 1 | MBLN |
表7
山荆子居群的分子变异分析"
| 变异来源 Source of variation | 自由度 df | 平方和 Sum of square | 变异组分 Variance component | 变异百分比 Percentage of variation | 概率值 P |
|---|---|---|---|---|---|
| 居群间Among population | 6 | 201.966 | 0.84873 | 6.97 | <0.001 |
| 居群内Within population | 206 | 2334.987 | 11.33487 | 93.03 | <0.001 |
| 总数 Total | 212 | 2536.948 | 12.18360 | 100 | <0.001 |
表8
6个来源地区山荆子群体间遗传分化系数"
| 6个山荆子群体 6 populations of Malus baccata | MBGS | MBHB | MBHLJ | MBJL | MBNM | MBSX |
|---|---|---|---|---|---|---|
| MBGS | 0.000 | |||||
| MBHB | 0.184 | 0.000 | ||||
| MBHLJ | 0.206 | 0.042 | 0.000 | |||
| MBJL | 0.216 | 0.035 | 0.002 | 0.000 | ||
| MBNM | 0.209 | 0.166 | 0.061 | 0.079 | 0.000 | |
| MBSX | -0.024 | 0.120 | 0.097 | 0.056 | 0.104 | 0.000 |
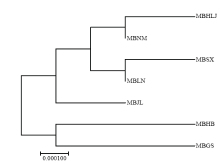
图2
基于群体间遗传距离构建的山荆子一致性NJ树"
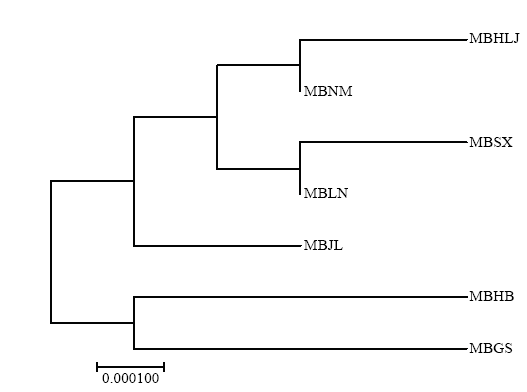
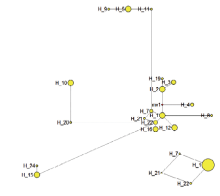
图3
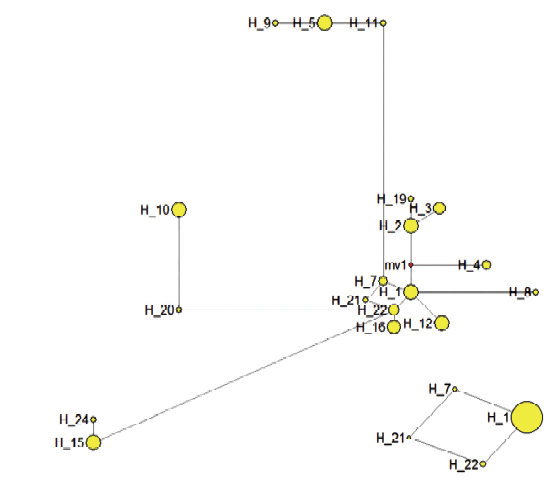
基于山荆子4个叶绿体基因间区合并序列的单倍型中介邻接网络图(Median-Joining network,MJ)"
| [1] | 中国植物志编辑委员会. 中国植物志[DB/OL]. [ 2019- 05- 17]. . |
| Editorial Committee of Flora Reipublicae Sinicae. Flora Reipublicae Popularis Sinicae [DB/OL]. [ 2019- 05- 17]. .(in Chinese) | |
| [2] | 俞德俊 . 中国果树分类学. 北京: 中国农业出版社, 1979: 91. |
| YU D J. Taxonomy of China Fruits. Beijing: China Agriculture Press, 1979: 91. (in Chinese) | |
| [3] | 钱关泽 . 苹果属 (Malus Mill.) 分类学研究[D]. 南京: 南京林业大学, 2005. |
| QIAN G Z . The taxonomic study of the genus Malus Mill.[D]. Nanjing: Nanjing Forestry University, 2005. ( in Chinese) | |
| [4] | ELITH J, GRAHAM C H, ANDERSON R P, DUDíK M, FERRIER S, GUISAN A, HIJMANS R J, HUETTMANN F, LEATHWICK J R, LEHMANN A, LI J, Lohmann L G, LOISELLE B A, MANION G, MORITZ C, NAKAMURA M, NAKAZAWA Y, OVERTON J M, PETERSON A T, PHILLIPS S J, RICHARDSON K, SCACCHETTI- PEREIRA R, SCHAPIRE R E, SOBERON J, WILLIAMS S, WISZ M S, ZIMMERMANN N E. Novel methods improve prediction of species’ distributions from occurrence data. Ecography, 2006,29(2):129-151. |
| [5] | 王大江, 王昆, 高源, 赵继荣, 刘立军, 龚欣, 李连文 . 我国苹果属资源现代分布调查初报. 植物遗传资源学报, 2017,18(6):1116-1124. |
| WANG D J, WANG K, GAO Y, ZHAO J R, LIU L J, GONG X, LI L W . Preliminary investigation of modern distribution of Malus resources in China. Journal of Plant Genetic Resources, 2017,18(6):1116-1124. (in Chinese) | |
| [6] | 杜学梅, 杨廷桢, 高敬东, 王骞, 蔡华成, 李春燕, 弓桂花 . 中国野生山定子的自然分布及利用研究现状. 中国农学通报, 2017,33(5):24-28. |
| DU X M, YANG T Z, GAO J D, WANG Q, CAI H C, LI C Y, GONG G H . Wild Malus baccata(L.) Borkh. in China: Natural distribution, utilization and study status. Chinese Agricultural Science Bulletin, 2017,33(5):24-28. (in Chinese) | |
| [7] | 王雷宏 . 山荆子(Malus baccata (L.) Borkh.)变异式样研究[D]. 南京: 南京林业大学, 2008. |
| WANG L H . A study on variation patterns in Malus baccata (L.) Borkh.[D]. Nanjing: Nanjing Forestry University, 2008. ( in Chinese) | |
| [8] | 陆秋农, 贾定贤 . 中国果树志•苹果卷. 北京: 中国林业出版社, 1999: 121. |
| LU Q N, JIA D X. Chinese Fruit Tree • Apple. Beijing: China Forestry Press, 1999: 121. (in Chinese) | |
| [9] | 李育农 . 苹果属植物种质资源研究. 北京: 中国农业出版社, 2001: 23-25. |
| LI Y N. Researches of Germplasm Resources of Malus Mill. Beijing: China Agriculture Press, 2001: 23-25. (in Chinese) | |
| [10] | 邹旭, 彭冶, 王璐, 李垚, 张往祥, 刘雪 . 末次盛冰期以来气候变化对中国山荆子分布格局的影响. 植物科学学报, 2018,36(5):676-686. |
| ZOU X, PENG Y, WANG L, LI Y, ZHANG W X, LIU X . Impact of climate change on the distribution pattern of Malus baccata(L.) Borkh. in China since the Last Glacial Maximum. Plant Science Journal, 2018,36(5):676-686. (in Chinese) | |
| [11] | 王雷宏, 汤庚国 . 山荆子腊叶标本表型性状变异分析. 西北植物学报, 2007,27(8):1690-1694. |
| WANG L H, TANG G G . Phenotypic variations of exsiccate- specimen of Malus baccata(L.) Borkh. Acta Botanica Boreali Occidentalia Sinica, 2007,27(8):1690-1694. (in Chinese) | |
| [12] | 王雷宏, 汤庚国, 夏海武, 蔡华 . 山荆子叶脉序的研究. 南京林业大学学报(自然科学版), 2008,32(2):39-42. |
| WANG L H, TANG G G, XIA H W, CAI H . Study on leaf venation of Malus baccata(L.) Borkh. Journal of Nanjing Forestry University (Natural Sciences Edition), 2008,32(2):39-42. (in Chinese) | |
| [13] | 陈曦, 汤庚国, 郑玉红, 王雷宏 . 苹果属山荆子遗传多样性的RAPD 分析. 西北植物学报, 2008,28(10):1954-1959. |
| CHEN X, TANG G G, ZHENG Y H, WANG L H . RAPD analysis of genetic diversity of Malus baccata. Acta Botanica Boreali Occidentalia Sinica, 2008,28(10):1954-1959. (in Chinese) | |
| [14] | ROBINSON J P, HARRIS S A, JUNIPER B E . Taxonomy of the genus Malus Mill.(Rosaceae ) with emphasis on the cultivated apple, Malus domestica Borkh. Plant Systematic and Evolution, 2001,226(1/2):35-58. |
| [15] | 王雷宏, 杨俊仙, 郑玉红, 汤庚国 . 苹果属山荆子地理分布模拟. 北京林业大学学报, 2011,33(3):70-73. |
| WANG L H, YANG J X, ZHENG Y H, TANG G G . Modelling the geographical distribution of Malus baccata. Journal of Beijing Forestry University, 2011,33(3):70-73. (in Chinese) | |
| [16] | 杨锋, 刘志, 伊凯, 刘延杰, 王强, 孙建设 . 东北山定子(Malus baccata (L.) Borkh.)野生居群表型遗传多样性分析及生态地理分布研究. 植物遗传资源学报, 2015,16(3):490-496. |
| YANG F, LIU Z, YI K, LIU Y J, WANG Q, SUN J S . Studies on geographical regions and analysis on genetic diversity of phenotypic of natural population of ‘Malus baccata ( L.) Borkh’ in Northeast of China. Journal of Plant Genetic Resources, 2015,16(3):490-496. (in Chinese) | |
| [17] | 王雷宏, 郑玉红, 汤庚国 . 8个山荆子居群遗传多样性的ISSR分析. 西北植物学报, 2010,30(7):1337-1343. |
| WANG L H, ZHENG Y H, TANG G G . ISSR analysis of genetic diversity of eight populations in Malus baccata. Acta Botanica Boreali Occidentalia Sinica, 2010,30(7):1337-1343. (in Chinese) | |
| [18] | 王雷宏, 郑玉红, 汤庚国 . 基于SSR标记的8个山荆子居群遗传多样性和遗传关系分析. 植物资源与环境学报, 2012,21(1):42-46. |
| WANG L H, ZHENG Y H, TANG G G . Analyses of genetic diversity and genetic relationship of eight populations of Malus baccata based on SSR marker. Journal of Plant Resources and Environment, 2012,21(1):42-46. (in Chinese) | |
| [19] | 高源, 王昆, 王大江, 赵继荣, 张彩霞, 丛佩华, 刘立军, 李连文, 朴继成 . 7 个来源地区山荆子的遗传多样性与群体结构分析. 中国农业科学, 2018,51(19):3766-3777. |
| GAO Y, WANG K, WANG D J, ZHAO J R, ZHANG C X, CONG P H, LIU L J, LI L W, PIAO J C . The genetic diversity and population structure analysis on Malus baccata(L.) Borkh. from 7 sources. Scientia Agricultura Sinica, 2018,51(19):3766-3777. (in Chinese) | |
| [20] | 倪梁红, 赵志礼, 米玛 . 药用植物叶绿体基因组研究进展. 中药材, 2015,38(9):1990-1994. |
| NI L H, ZHAO Z L, MI M . Advances in research on chloroplast genome of medicinal plants. Journal of Chinese Medicinal Materials, 2015,38(9):1990-1994. (in Chinese) | |
| [21] | 李宏韬, 赵淑青, 赵彦修, 张慧 . 叶绿体基因工程简介. 遗传, 2003,25(4):495-498. |
| LI H T, ZHAO S Q, ZHAO X X, ZHANG H . The introduction of chloroplast gene engineering. Hereditas, 2003,25(4):495-498. (in Chinese) | |
| [22] | 张韵洁, 李德铢 . 叶绿体系统发育基因组学的研究进展. 植物分类与资源学报, 2011,33(4):365-375. |
| ZHANG Y J, ZHANG D Z . Advances in phylogenomics based on complete chloroplast genomes. Plant Diversity and Resources, 2011,33(4):365-375. (in Chinese) | |
| [23] | 付涛, 王志龙, 钱萍仙, 李文, 袁冬明, 严春风 . 高等植物DNA条形码最新研究进展及其应用. 核农学报, 2016,30(5):887-896. |
| FU T, WANG Z L, QIAN P X, LI W, YUAN D M, YAN C F . The latest research progress and application of the DNA barcode in higher plants. Journal of Nuclear Agricultural Sciences, 2016,30(5):887-896. (in Chinese) | |
| [24] | SAVOLAINEN V, CORBAZ R, MONCOUSIN C, SPICHIPER R, MANEN J F . Chloroplast DNA variation and parentage analysis in 55 apples. Theoretical and Applied Genetics, 1995,90:1138-1141. |
| [25] | ROBINSON J P, HARRIS S A, JUNIPER B E . Taxonomy of the genus Malus Mill. (Rosaceae) with emphasis on the cultivated apple, Malus domestica Borkh. Plant Systematics and Evolution, 2001,226:35-58. |
| [26] | COART E, VAN GLABEKE S, DE LOOSE M, LARSEN A S, ROLDAN-RUIZ I . Chloroplast diversity in the genus Malus: new insights into the relationship between the European wild apple (Malus sylvestris (L) Mill.) and the domesticated apple (Malus domestica Borkh.). Molecular Ecology, 2006,15:2171-2182. |
| [27] | NIKIFOROVA S V, CAVALIERI D, VELASCO R, GOREMYKIN V . Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Molecular Biology Evolution, 2013,30(8):1751-1760. |
| [28] | 丁芳兵. 湖北海棠(Malus.hupehensis)及近缘种的matK和ITS序列分析[D]. 南京: 南京林业大学, 2012. |
| DING F B . Sequence analysis of matK and ITS between Malus hupehensis and its relative species[D]. Nanjing: Nanjing Forestry University, 2012. ( in Chinese) | |
| [29] | 朱元娣, 曹敏格, 许正, 王昆, 张文 . 基于ITS和matK序列探讨新疆野苹果与中国苹果的系统演化关系. 园艺学报, 2014,41(2):227-239. |
| ZHU Y D, CAO M G, XU Z, WANG K, ZHANG W . Phylogenetic relationship between Xinjiang wild apple(Malus sieversii Roem.)and Chinese apple(Malus × domestica subsp. chinensis)based on ITS and matK sequences. Acta Horticulturae Sinica, 2014,41(2):227-239. (in Chinese) | |
| [30] | 徐榕雪 . 基于分子标记技术的部分苹果属植物分类地位及亲缘关系研究[D]. 南京: 南京林业大学, 2018. |
| XU R X . Taxonomic status and phylogenetic relationships of some Malus plants based on molecular marker technology[D]. Nanjing: Nanjing Forestry University, 2018. ( in Chinese) | |
| [31] | 李慧峰 . 泰沂山区苹果属植物系统学研究[D]. 沈阳: 沈阳农业大学, 2012. |
| LI H F . Studies on the taxonomy of the genus Malus Mill. (Rosaceae) of Taiyi-Mountains[D]. Shenyang: Shenyang Agricultural University, 2012. ( in Chinese) | |
| [32] | BARAKET G, OLFA S, KHALED C, MESSAOUD M, MOHAMED M, MOKHTAR T, AMEL S H . Chloroplast DNA analysis in Tunisian fig cultivars (Ficus carica L.): Sequence variations of the trnl-trnf intergenic spacer. Biochemical Systematics and Ecology, 2009,36(11):828-835. |
| [33] | POTTER D, LUBY J J, HARRISON R E . Phylogenetic relationships among species of Fragaria (Rosaceae) inferred from non-coding nuclear and chloroplast DNA sequences. Systematic Botany, 2009,25(2):337-348. |
| [34] | GHADA B, AHMED B A, KHALED C, OLFA S, MESSAOUD M, MOKHTAR T, AMEL S H . Molecular evolution of chloroplast DNA in fig (Ficus carica L.): Footprints of sweep selection and recent expansion. Biochemical Systematics and Ecology, 2010,38(4):563-575. |
| [35] | SHAW J, LICKEY E B, BECK J T, FARMER S B, LIU W S, MILLER J, SIRIPUN K C, WINDER C T, SCHILLING E E, SMALL R L . The tortoise and the hare Ⅱ: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. American Journal of Botany, 2005,921(1):142-166. |
| [36] | VOLK G M, HENK A D, BALDO A, FAZIO G, CHAO C T, RICHARDS C M . Chloroplast heterogeneity and historical admixture within the genus Malus. American Journal of Botany, 2015,102(7):1198-1208. |
| [37] | KUMAR S, STECHER G, TAMURA K . MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology Evolution, 2016,33(7):1870-1874. |
| [38] | LIBRADO P, ROZAS J . DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 2009,25:1451-1452. |
| [39] | SAITO N, NEI M . The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 1987,4(4):406-425. |
| [40] | EXCOFFIER L, LISCHER H E L . Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 2010,10:564-567. |
| [41] | KIMURA T, IKETANI H, KOTOBUKI K, MATSUTA N, BAN Y, HAYASHI T, YAMAMOTO T . Genetic characterization of pear varieties revealed by chloroplast DNA sequences. Journal of Horticultural Science and Biotechnology, 2003,7:241-247. |
| [42] | PETIT R, AGUINAGALDE I, DE BEAULIEU J, BITTKAU C, BREWER S, CHEDDADI R, ENNOS R, FINESCHI S, GRIVET D, LASCOUX M, MOHANTY A, MÜLLER-STARCK G, DEMESURE- MUSCH B, PALMÉ A, MARTÍN J P, RENDELL S, VENDRAMIN G G . Glacial refugia: Hotspots but not melting pots of genetic diversity. Science, 2003,300:1563-1565. |
| [43] | KATAYAMA H, OGIHARA Y . Phylogenetic affinities of the grasses to other monocots as revealed by molecular analysis of chloroplast DNA. Current Genetics, 1996,20:527-581. |
| [44] | EIADTHONG W, YONEMORI K, SUGIURA A, UTSUNOMIYA N, SUBHADRABANDHU S . Analysis of phylogenetic relationships in Mangifera by restriction site analysis of an amplified region of cpDNA. Scientia Horticulturae, 1999,80:145-150. |
| [45] | LIU J, ZHENG X Y, DANEIL P, HU C Y, TENG Y W . Genetic diversity and population structure of Pyrus Calleryana (Rosaceae) in Zhejiang province, China. Biochemical Systematics and Ecology, 2012,45:69-78. |
| [46] | 宗宇 . 中国北方野生杜梨的遗传多样性和谱系地理研究[D]. 杭州: 浙江大学, 2014. |
| ZONG Y . Studies on genetic diversity and phylogeography of wild Pyrus betulaefolia in Northern China[D]. Hangzhou: Zhejiang University, 2014. ( in Chinese) | |
| [47] | NEI M . Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 1978,89(3):583-590. |
| [48] | AVISE J C, ARNOLD J, BALL R M, BERMINGHAM E, LAMB T, NEIGEL J E, REEB C A, SAUNDER N C . Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annual Review of Ecology and Systematics, 1987,18:489-522. |
| [49] | WRIGHT S. Evolution and the Genetics of Populations, Variability Within and Among Natural Populations. Chicago: The University of Chicago Press, 1978: 4. |
| [50] | ZHANG C Y, CHEN X S, HE T M, LIU X L, FENG T, YUAN Z H . Genetic structure of Malus sieversii population from Xinjiang, China, revealed by SSR markers. Journal of Genetics and Genomics, 2007,34(10):947-955. |
| [51] | POSADA D, CRANDALL K A . Intraspecific gene genealogies: Trees grafting into networks. Trends in Ecology and Evolution, 2001,16:37-45. |
| [1] | 姜朋, 张鹏, 姚金保, 吴磊, 何漪, 李畅, 马鸿翔, 张旭. 宁麦系列小麦品种的性状特点及相关基因位点分析[J]. 中国农业科学, 2022, 55(2): 233-247. |
| [2] | 李晓川,王朝海,周平,马维,吴瑞,宋治豪,梅艳. 马铃薯品种(系)田间晚疫病抗性评价和全基因组遗传多样性分析[J]. 中国农业科学, 2022, 55(18): 3484-3500. |
| [3] | 万映伶,朱梦婷,刘爱青,金亦佳,刘燕. 中国观赏芍药表型多样性解析与资源评价[J]. 中国农业科学, 2022, 55(18): 3629-3639. |
| [4] | 胡光明,张琼,韩飞,李大卫,李作洲,汪志,赵婷婷,田华,刘小莉,钟彩虹. 猕猴桃属植物通用型SSR分子标记引物的筛选及应用[J]. 中国农业科学, 2022, 55(17): 3411-3425. |
| [5] | 王璐伟,沈志军,李贺欢,潘磊,牛良,崔国朝,曾文芳,王志强,鲁振华. 基于SSR荧光标记的79份桃种质遗传多样性分析[J]. 中国农业科学, 2022, 55(15): 3002-3017. |
| [6] | 陈旭,郝雅琼,聂兴华,杨海莹,刘松,王雪峰,曹庆芹,秦岭,邢宇. 板栗总苞和坚果主要性状与SSR标记的关联分析[J]. 中国农业科学, 2022, 55(13): 2613-2628. |
| [7] | 徐晓,任根增,赵欣蕊,常金华,崔江慧. 中国高粱地方品种和育成品种穗部表型性状精准鉴定及综合评价[J]. 中国农业科学, 2022, 55(11): 2092-2108. |
| [8] | 唐修君,樊艳凤,贾晓旭,葛庆联,陆俊贤,唐梦君,韩威,高玉时. 基于线粒体DNA D-loop区的肉鸡品种遗传多样性和起源分析[J]. 中国农业科学, 2021, 54(24): 5302-5315. |
| [9] | 李昕芫, 娄金秀, 刘清源, 胡健, 张英俊. 中国东北和华北地区紫花苜蓿根瘤菌遗传多样性研究[J]. 中国农业科学, 2021, 54(16): 3393-3405. |
| [10] | 王富强,张建,温常龙,樊秀彩,张颖,孙磊,刘崇怀,姜建福. 基于KASP标记的葡萄品种鉴定[J]. 中国农业科学, 2021, 54(13): 2830-2842. |
| [11] | 杨涛,黄雅婕,李生梅,任丹,崔进鑫,庞博,于爽,高文伟. 海岛棉种质资源表型性状的遗传多样性分析及综合评价[J]. 中国农业科学, 2021, 54(12): 2499-2509. |
| [12] | 崔一平,彭埃天,宋晓兵,程保平,凌金锋,陈霞. 广东梅州柑橘黄龙病和病毒病的发生调查及其黄龙病菌原噬菌体多样性[J]. 中国农业科学, 2020, 53(8): 1572-1582. |
| [13] | 常佳迎,刘树森,石洁,郭宁,张海剑,马红霞,杨春凤. 海南三亚和黄淮海地区玉米小斑病菌致病性及遗传多样性分析[J]. 中国农业科学, 2020, 53(6): 1154-1165. |
| [14] | 徐默然,蔺瑞明,王凤涛,冯晶,徐世昌. 103份小麦品种(系)抗条锈性和遗传多样性评价及基因检测[J]. 中国农业科学, 2020, 53(4): 748-760. |
| [15] | 王宝宝,郭成,孙素丽,夏玉生,朱振东,段灿星. 玉米穗腐病致病禾谷镰孢复合种的遗传多样性、致病力与毒素化学型分析[J]. 中国农业科学, 2020, 53(23): 4777-4790. |
|
||