






中国农业科学 ›› 2025, Vol. 58 ›› Issue (6): 1065-1082.doi: 10.3864/j.issn.0578-1752.2025.06.003
潘丽媛1( ), 王永军1, 李海军1, 侯富1, 李菁1, 李丽丽1, 孙苏阳1,2,3()
), 王永军1, 李海军1, 侯富1, 李菁1, 李丽丽1, 孙苏阳1,2,3()
收稿日期:2024-08-04
接受日期:2024-09-20
出版日期:2025-03-16
发布日期:2025-03-25
通信作者:
联系方式:
潘丽媛,E-mail:panly89@126.com。
基金资助:
PAN LiYuan1(), WANG YongJun1, LI HaiJun1, HOU Fu1, LI Jing1, LI LiLi1, SUN SuYang1,2,3()
Received:2024-08-04
Accepted:2024-09-20
Published:2025-03-16
Online:2025-03-25
摘要:
【目的】 小麦作为重要的粮食作物,鉴定参与籽粒蛋白质合成的基因调控网络,并确定关键候选基因,为小麦品质育种和改良提供理论依据。【方法】 以淮麦48的6个发育时期(花后5、10、15、20、25和30 d)的籽粒为研究材料,总结小麦籽粒蛋白质累积规律,利用WGCNA方法分析转录组数据和籽粒蛋白质含量表型数据,构建加权基因共表达网络,筛选关键枢纽转录因子(transcript factors,TFs)。【结果】 小麦籽粒蛋白质含量积累过程表现为先减少后升高的趋势,并且在花后25 d达到最低值(12.16%),相邻发育时期的蛋白质含量差异显著。在相邻发育时期之间共鉴定到25 427个差异表达基因(differentially expressed genes,DEGs)。通过聚类分析可分为5组(A—E),其中,B组的差异表达基因数量最多(10 906个),包含49个家族的1 022个转录因子,其中,NAC家族的TFs数量最多(107个)。通过WGCNA分析,发现5个与蛋白质含量显著相关的共表达模块。turquoise模块与蛋白质含量的正相关性最高(r=0.80,P=1×10-4)。通过整合差异表达基因和加权基因共表达网络,在2个模块(turquoise和blue)中发现了6个正向调控的枢纽TFs,分别来自MIKC-MADS、TCP、TALE和CPP家族。进一步对淮麦48的蛋白质含量与不同时期的基因表达量进行相关性分析,发现其中5个枢纽TFs的表达量与蛋白质含量存在显著正相关性,并且TraesCS5B03G0740100和TraesCS7D03G0590500在穗和籽粒中特异性高表达。【结论】 鉴定到与小麦蛋白质含量累积相关的重要模块turquoise和blue模块,筛选到6个枢纽TFs,并发现2个重要的枢纽基因表达量与蛋白质含量呈显著正相关,且在穗和籽粒组织中特异性高表达,可作为小麦籽粒蛋白质积累调控的候选基因。
潘丽媛, 王永军, 李海军, 侯富, 李菁, 李丽丽, 孙苏阳. 基于转录组和WGCNA筛选小麦籽粒蛋白质累积相关调控基因[J]. 中国农业科学, 2025, 58(6): 1065-1082.
PAN LiYuan, WANG YongJun, LI HaiJun, HOU Fu, LI Jing, LI LiLi, SUN SuYang. Screening Regulatory Genes Related to Wheat Grain Protein Accumulation Based on Transcriptome and WGCNA Analysis[J]. Scientia Agricultura Sinica, 2025, 58(6): 1065-1082.
表1
本研究用于实时荧光定量PCR的引物序列"
| 基因ID Gene ID | 正向引物 Forward primer (5′-3′) | 反向引物 Reverse primer (5′-3′) |
|---|---|---|
| TraesCS3A03G0074600 | CGGAGATCATTGCGTAGGG | GGAATTAGCACGTGTGGCG |
| TraesCS3A03G0885700 | TCTTCCTTCATCGCTTCCATC | TCCTCAGGGTGCCATCTATCA |
| TraesCS3A03G0888300 | TACCCAGTTTTAGTAGACGCCC | CTCGTTTTGTCCCAGTGTGC |
| TraesCS3A03G0891600 | GAAGTGCGACGTGGATATAAGG | GAAGGTACTGAGAGAGGCGAGA |
| TraesCS3A03G0891900 | GAAGGCTTTTGGGGATGCT | GCTATCCAATCGCTCTCCTCTT |
| TraesCS3A03G0892500 | TCGTTCGGATGACAGTGCTT | CTATCCCACATTTGGAAACAGG |
| TraesCS4B03G0043800 | GAACAAATCCCAGCCATCCA | ACGAGTAGGCGTTGGGTATTC |
| TraesCS4B03G0823800 | TCCACCAGATGGCGGAGTT | GGCGTGGGGAAGAGCGT |
| TraesCS6B03G0310500 | GGGGAGGCACTATTGCTTTC | CCAAAGTAGTCCGAAGCGTATC |
| TraesCS6B03G0312000 | CCTGCTTTGATGGCTGGAC | AAGAAGGGGAATGGGGCT |
| TaActin | GGAATGGTCAAGGCTGGTTT | CGGAGCTCGTTGTAGAATGTGT |
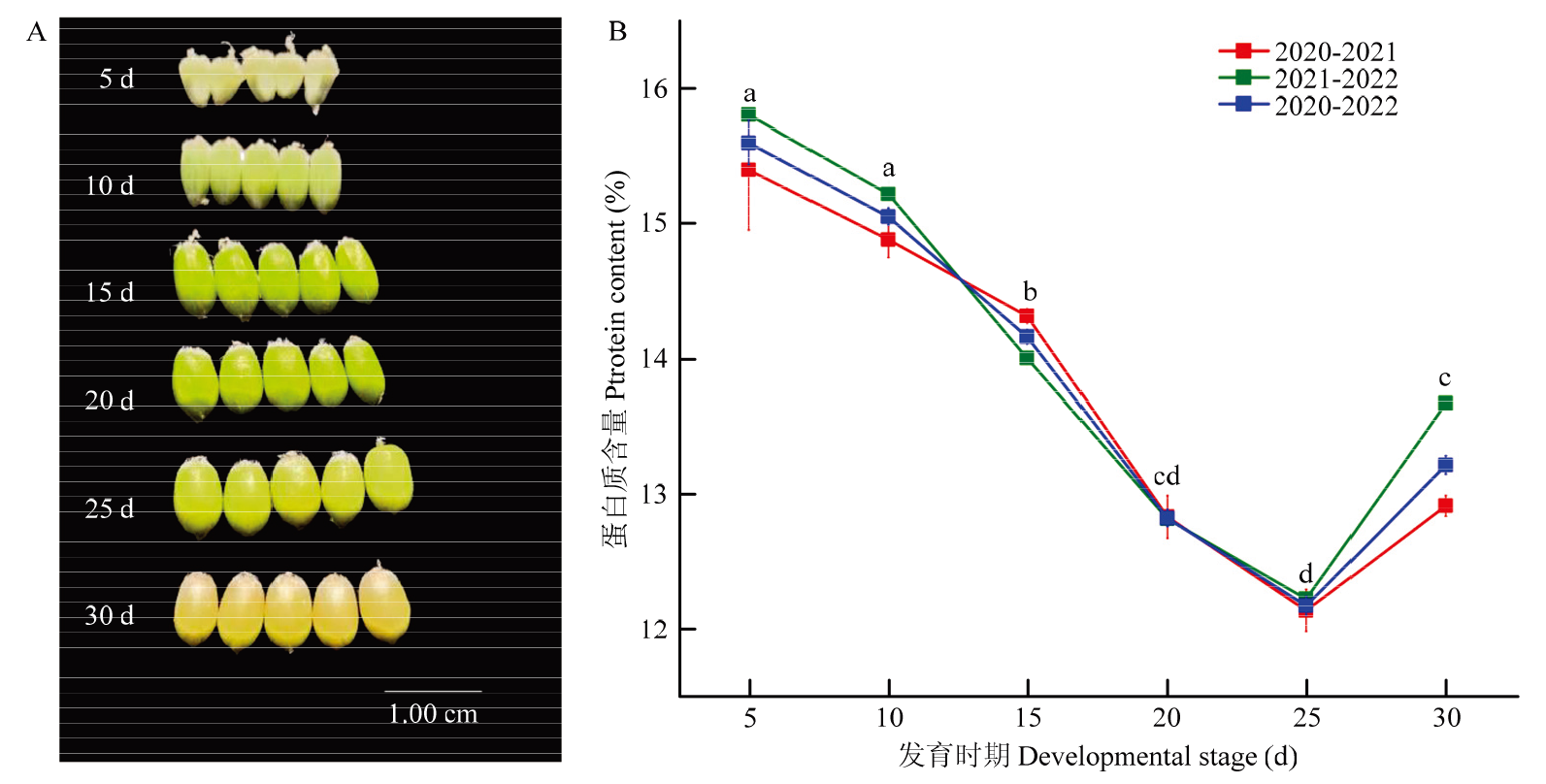
图1
淮麦48籽粒不同发育时期的蛋白质含量变化 A:6个不同发育时期淮麦48的籽粒;B:6个不同发育时期淮麦48籽粒蛋白质含量变化图。不同字母代表在0.05水平差异显著"
表2
不同发育时期淮麦48样本的转录组测序质量"
| 样本 Sample | 时期 Stage (d) | 原始reads数 Raw reads | 质控后reads数 Saved reads | 质量分数 High quality rate (%) | 比对率 Reads aligned (%) | 多次比对率 Multialignment (%) | 单一比对率 Unique alignment (%) |
|---|---|---|---|---|---|---|---|
| 05D.Rep1 | 5 | 53494726 | 53253286 | 99.55 | 88.31 | 6.18 | 94.49 |
| 05D.Rep2 | 5 | 41144322 | 40944134 | 99.51 | 88.17 | 6.22 | 94.38 |
| 05D.Rep3 | 5 | 42043100 | 41848014 | 99.54 | 88.55 | 6.14 | 94.69 |
| 10D.Rep1 | 10 | 43073974 | 42750556 | 99.25 | 82.85 | 6.39 | 89.23 |
| 10D.Rep2 | 10 | 45712962 | 45445466 | 99.41 | 85.54 | 6.37 | 91.91 |
| 10D.Rep3 | 10 | 47715540 | 47273162 | 99.07 | 80.03 | 6.62 | 86.65 |
| 15D.Rep1 | 15 | 46401552 | 45997986 | 99.13 | 73.62 | 8.65 | 82.27 |
| 15D.Rep2 | 15 | 40517866 | 40147604 | 99.09 | 72.56 | 8.86 | 81.42 |
| 15D.Rep3 | 15 | 44238716 | 43790148 | 98.99 | 71.59 | 9.02 | 80.61 |
| 20D.Rep1 | 20 | 37942654 | 37577108 | 99.04 | 70.18 | 10.93 | 81.11 |
| 20D.Rep2 | 20 | 41530148 | 41087334 | 98.93 | 68.35 | 11.76 | 80.11 |
| 20D.Rep3 | 20 | 53574728 | 52925588 | 98.79 | 69.67 | 14.02 | 83.69 |
| 25D.Rep1 | 25 | 52320432 | 51672172 | 98.76 | 66.29 | 14.04 | 80.34 |
| 25D.Rep2 | 25 | 46626444 | 46097376 | 98.87 | 66.08 | 13.96 | 80.04 |
| 25D.Rep3 | 25 | 51864528 | 51434414 | 99.17 | 69.34 | 12.86 | 82.20 |
| 30D.Rep1 | 30 | 54570678 | 53738308 | 98.47 | 61.09 | 16.36 | 77.44 |
| 30D.Rep2 | 30 | 51852354 | 51090038 | 98.53 | 67.55 | 12.04 | 79.59 |
| 30D.Rep3 | 30 | 55777132 | 55100978 | 98.79 | 68.56 | 12.38 | 80.94 |
| 共计/平均Total/Mean | 850401856 | 842173672 | 99.05 | 74.35 | 10.16 | 84.51 |
表3
淮麦48不同样本检测到的基因数"
| 样本 Sample | 已知基因数 No. of known genes | 新基因数 No. of new genes | 所有基因数 No. of all genes |
|---|---|---|---|
| 05D.Rep1 | 87963 | 6413 | 94376 |
| 05D.Rep2 | 86713 | 6512 | 93225 |
| 05D.Rep3 | 86541 | 6394 | 92935 |
| 10D.Rep1 | 84695 | 6534 | 91229 |
| 10D.Rep2 | 87409 | 6613 | 94022 |
| 10D.Rep3 | 84971 | 6509 | 91480 |
| 15D.Rep1 | 83938 | 6406 | 90344 |
| 15D.Rep2 | 80963 | 6210 | 87173 |
| 15D.Rep3 | 81302 | 6156 | 87458 |
| 20D.Rep1 | 78899 | 6029 | 84928 |
| 20D.Rep2 | 78802 | 5994 | 84796 |
| 20D.Rep3 | 81241 | 6308 | 87549 |
| 25D.Rep1 | 79118 | 6076 | 85194 |
| 25D.Rep2 | 79869 | 6076 | 85945 |
| 25D.Rep3 | 82747 | 6337 | 89084 |
| 30D.Rep1 | 77776 | 5945 | 83721 |
| 30D.Rep2 | 81517 | 6167 | 87684 |
| 30D.Rep3 | 80690 | 6187 | 86877 |
| 平均Mean | 82509 | 6270 | 88779 |
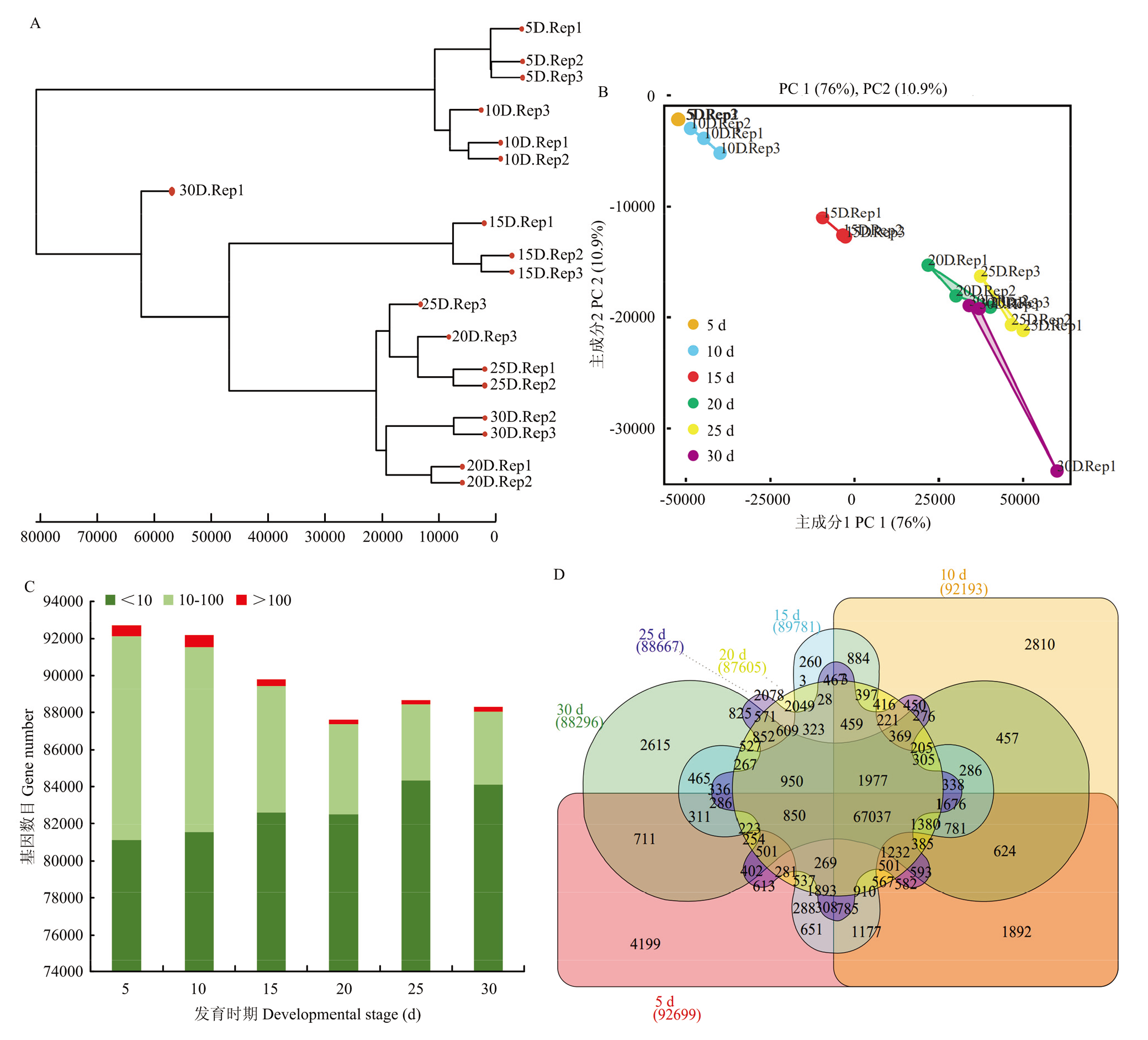
图2
淮麦48籽粒不同发育时期中基因的表达谱 A:6个不同发育时期和不同重复基因表达的聚类分析图;B:6个不同发育时期样本的基因表达量主成分分析图;C:每个发育时期的基因表达量分类图;D:6个不同发育时期样本中表达基因数目的韦恩图"
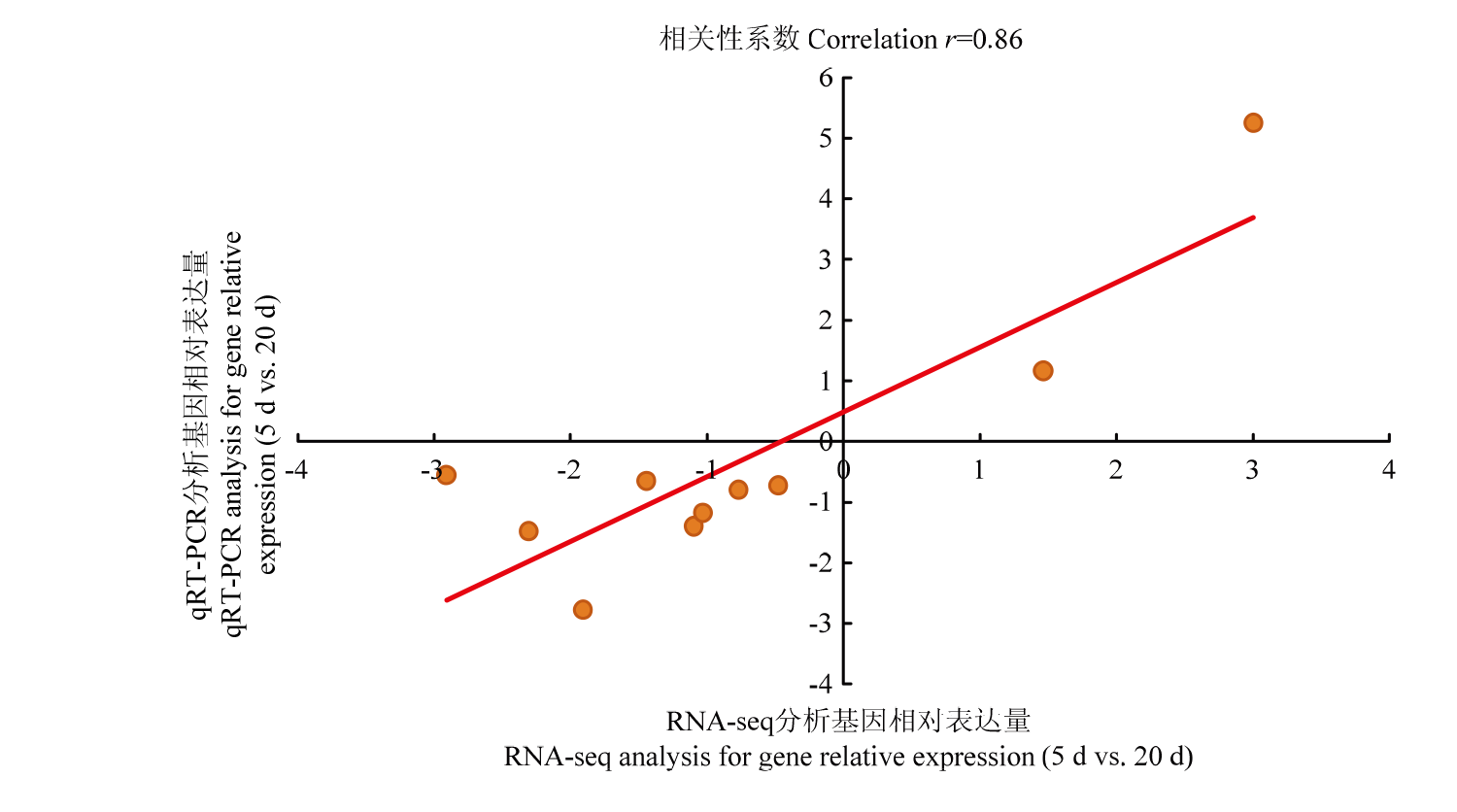
图3
转录组和qRT-PCR检测不同基因表达量的相关性分析"
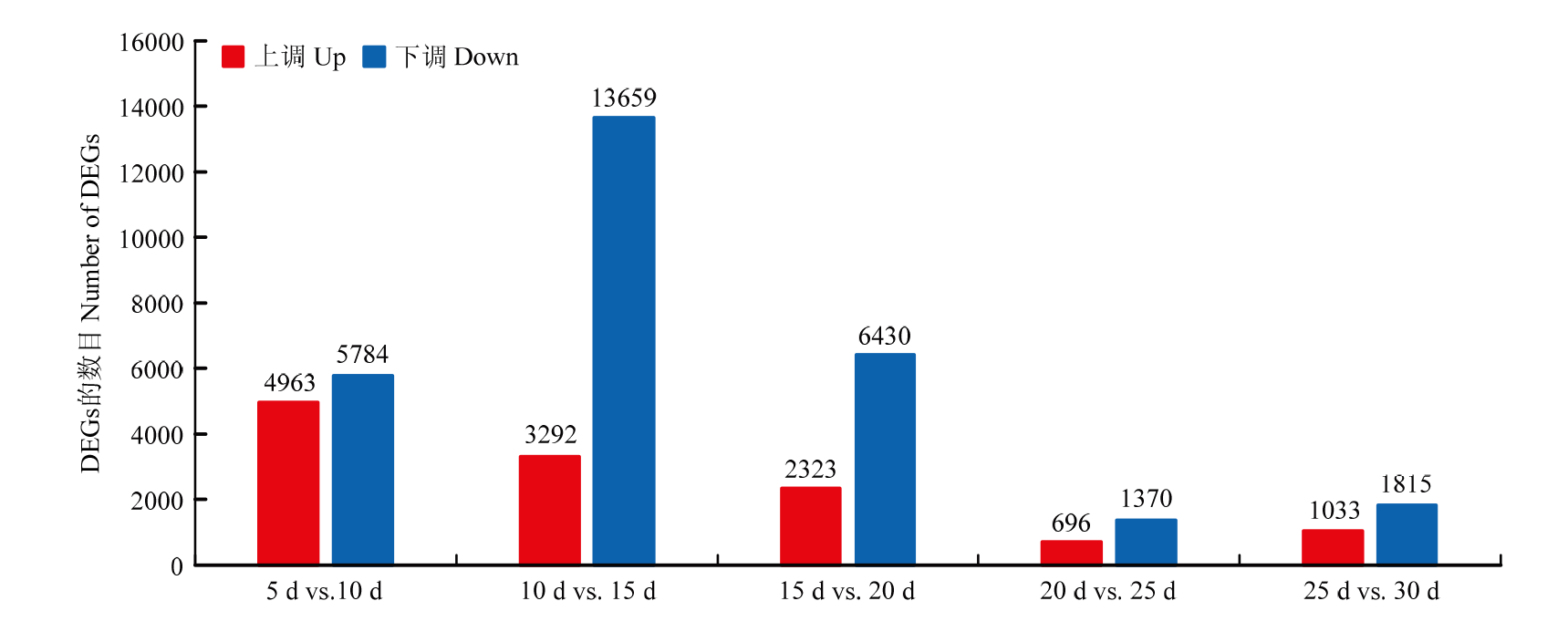
图4
淮麦48籽粒不同发育时期之间的差异表达基因"
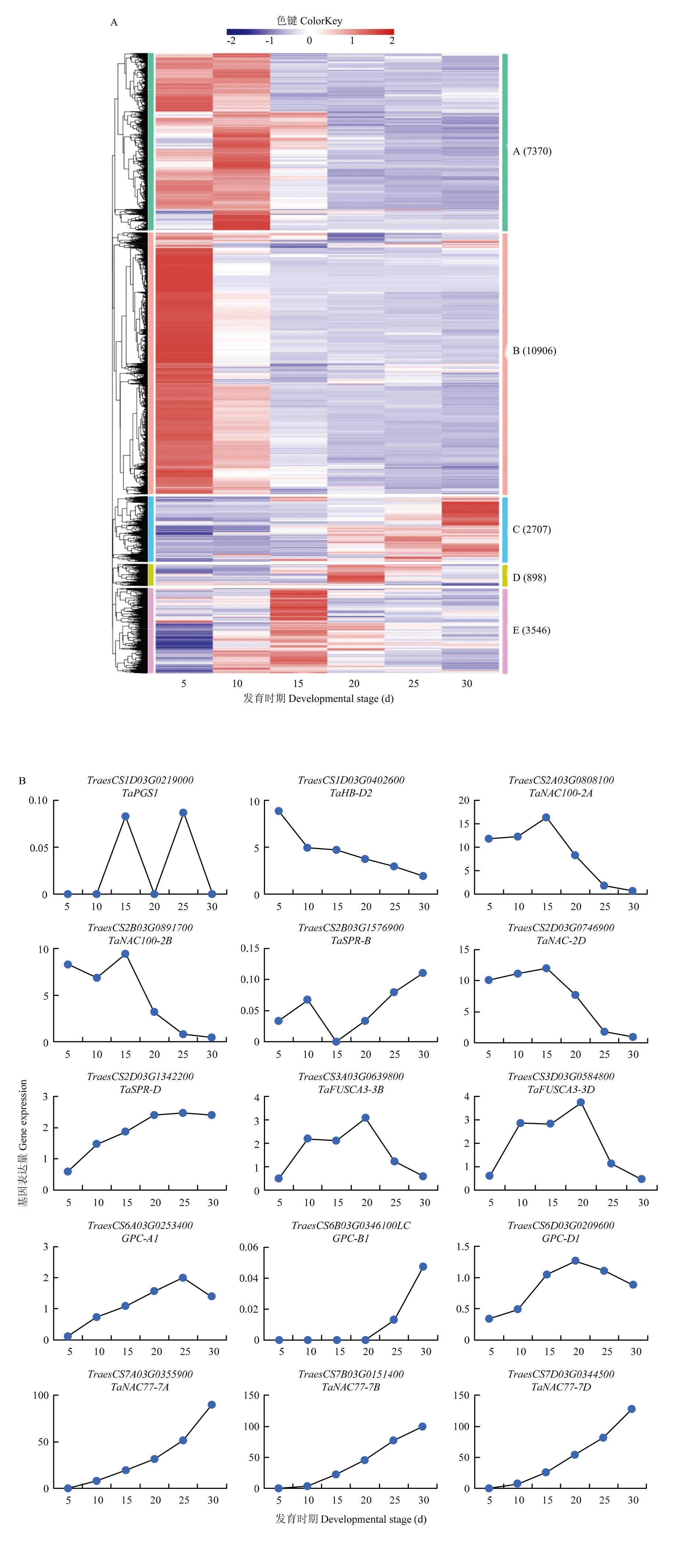
图5
淮麦48籽粒中所有DEGs的聚类分析及已知基因的表达谱 A:淮麦48籽粒6个不同发育时期的差异表达基因聚类分析;B:淮麦48中已报道的相关基因的表达模式"
表4
转录因子的分类及表达模式"
| 家族 Family | 数目 Number | 5种表达模式 Five expression patterns | 家族 Family | 数目 Number | 5种表达模式 Five expression patterns | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | A | B | C | D | E | |||||
| AP2 | 22 | 4 | 8 | 9 | 1 | 0 | HRT-like | 1 | 0 | 1 | 0 | 0 | 0 | |
| ARF | 14 | 4 | 9 | 1 | 0 | 0 | HSF | 24 | 5 | 1 | 15 | 0 | 3 | |
| ARR-B | 11 | 1 | 9 | 0 | 0 | 1 | LBD | 17 | 5 | 5 | 0 | 0 | 7 | |
| B3 | 49 | 4 | 30 | 2 | 5 | 8 | LSD | 1 | 0 | 1 | 0 | 0 | 0 | |
| BES1 | 2 | 1 | 1 | 0 | 0 | 0 | MIKC_MADS | 37 | 1 | 32 | 0 | 2 | 2 | |
| bHLH | 75 | 14 | 38 | 8 | 4 | 11 | M-type_MADS | 11 | 2 | 3 | 1 | 0 | 5 | |
| bZIP | 59 | 12 | 19 | 10 | 3 | 15 | MYB | 90 | 22 | 34 | 8 | 2 | 24 | |
| C2H2 | 37 | 9 | 18 | 5 | 2 | 3 | MYB_related | 28 | 14 | 9 | 1 | 2 | 2 | |
| C3H | 21 | 7 | 10 | 3 | 1 | 0 | NAC | 107 | 28 | 22 | 33 | 3 | 21 | |
| CAMTA | 1 | 0 | 1 | 0 | 0 | 0 | NF-YA | 10 | 0 | 0 | 0 | 4 | 6 | |
| CO-like | 3 | 0 | 2 | 0 | 0 | 1 | NF-YB | 14 | 6 | 4 | 0 | 0 | 4 | |
| CPP | 3 | 2 | 1 | 0 | 0 | 0 | NF-YC | 9 | 1 | 8 | 0 | 0 | 0 | |
| DBB | 8 | 2 | 4 | 0 | 0 | 2 | Nin-like | 3 | 3 | 0 | 0 | 0 | 0 | |
| Dof | 14 | 1 | 4 | 3 | 1 | 5 | S1Fa-like | 1 | 1 | 0 | 0 | 0 | 0 | |
| E2F/DP | 12 | 5 | 5 | 0 | 0 | 2 | SBP | 11 | 0 | 10 | 0 | 0 | 1 | |
| EIL | 4 | 2 | 2 | 0 | 0 | 0 | SRS | 1 | 0 | 0 | 1 | 0 | 0 | |
| ERF | 47 | 20 | 11 | 4 | 4 | 8 | TALE | 13 | 3 | 7 | 2 | 1 | 0 | |
| FAR1 | 15 | 2 | 3 | 3 | 0 | 7 | TCP | 22 | 1 | 21 | 0 | 0 | 0 | |
| G2-like | 47 | 20 | 17 | 2 | 1 | 7 | Trihelix | 22 | 6 | 10 | 1 | 0 | 5 | |
| GATA | 4 | 0 | 2 | 0 | 2 | 0 | Whirly | 6 | 1 | 5 | 0 | 0 | 0 | |
| GeBP | 2 | 1 | 1 | 0 | 0 | 0 | WOX | 9 | 4 | 1 | 3 | 1 | 0 | |
| GRAS | 24 | 8 | 11 | 1 | 2 | 2 | WRKY | 34 | 4 | 14 | 1 | 4 | 11 | |
| GRF | 7 | 0 | 6 | 0 | 1 | 0 | YABBY | 9 | 0 | 7 | 2 | 0 | 0 | |
| HB-other | 3 | 0 | 2 | 0 | 0 | 1 | ZF-HD | 11 | 2 | 3 | 6 | 0 | 0 | |
| HD-ZIP | 47 | 7 | 35 | 3 | 1 | 1 | 总计Total | 1022 | 235 | 447 | 128 | 47 | 165 | |
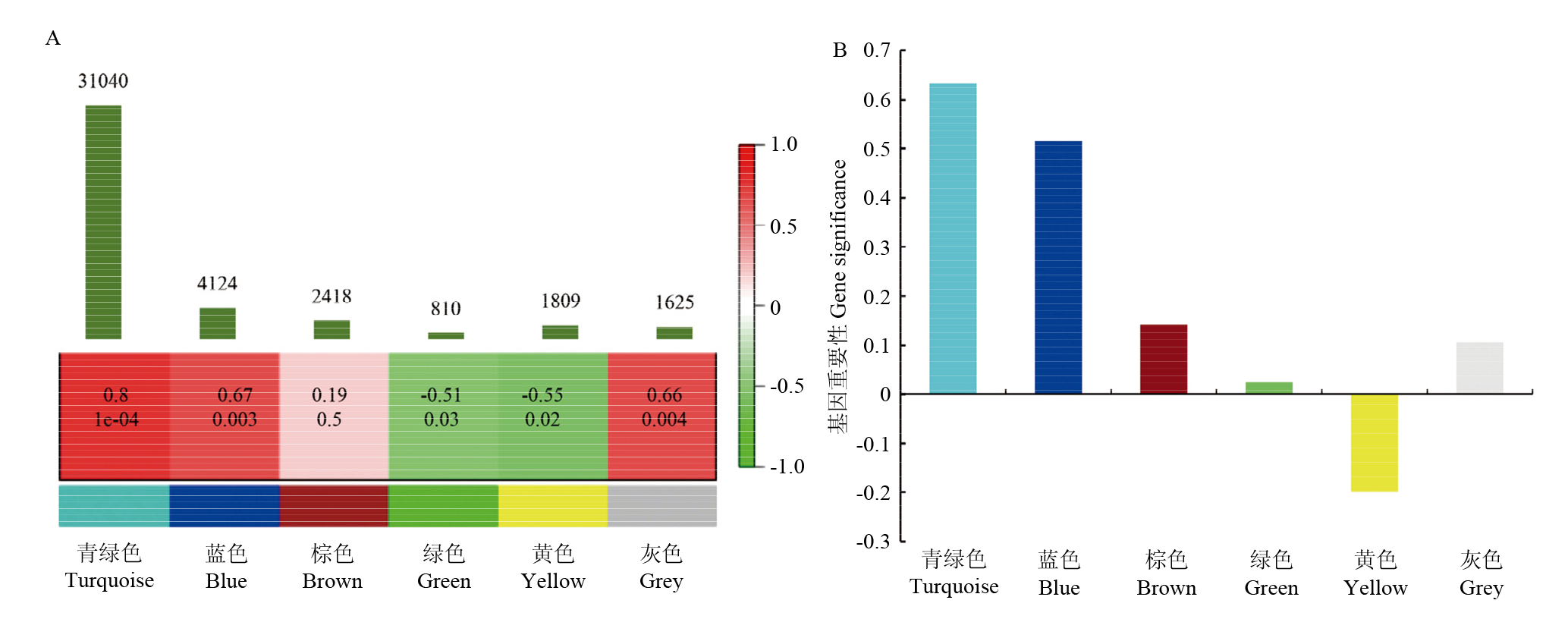
图6
加权基因共表达网络与蛋白质含量的相关性 A:基因模块与蛋白质含量之间的相关性分析。由下而上第一行表示WGCNA模块。第二行表示该模块与蛋白质含量的相关性,2个数字分别表示相关性系数及P值。最上方的柱状图表示每个模块含有的基因数目;B:不同模块基因显著性值分布图"
表5
小麦籽粒蛋白质含量调控的候选TFs"
| 基因ID Gene ID | 类别Description | 基因连通性值kME | 所属模块The modlue |
|---|---|---|---|
| TraesCS2A03G0362900 | MADS-box转录因子 MADS-box transcription factor | 0.986 | 青绿色 Turquoise |
| TraesCS4D03G0036300 | TALE转录因子 TALE transcription factor | 0.981 | 青绿色 Turquoise |
| TraesCS5A03G0079200 | TCP转录因子 TCP transcription factor | 0.984 | 青绿色 Turquoise |
| TraesCS5B03G0740100 | MADS-box转录因子 MADS-box transcription factor | 0.987 | 青绿色 Turquoise |
| TraesCS7D03G0590500 | MADS-box转录因子 MADS-box transcription factor | 0.981 | 青绿色 Turquoise |
| TraesCS5A03G0135400 | CPP转录因子 CPP transcription factor | 0.981 | 蓝色 Blue |
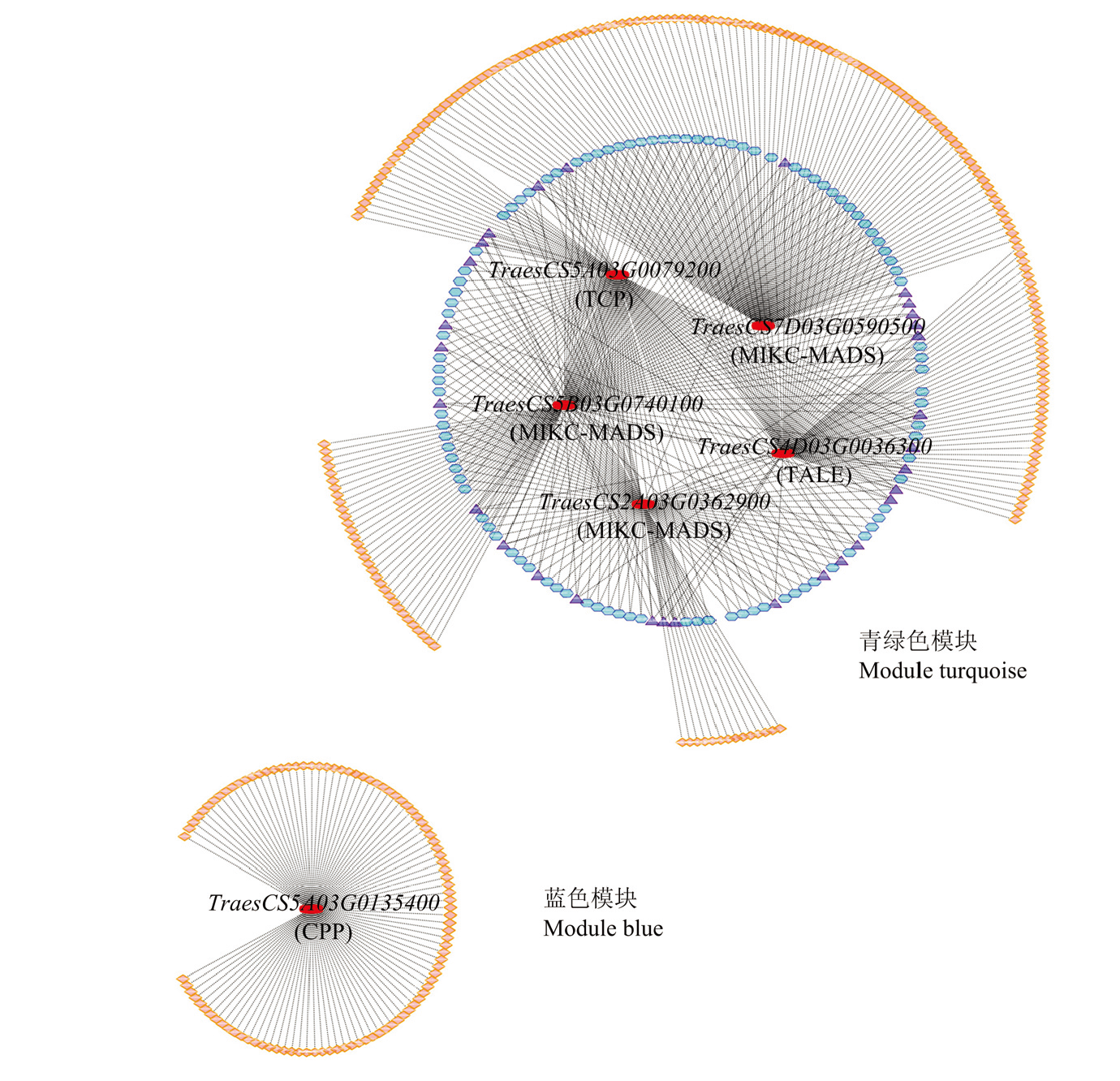
图7
加权基因共表达网络中的枢纽转录因子 图中展示为正向调控模块且kME>0.98的前100个基因。节点的大小表示调控基因的数目,红色节点表示hub转录因子。图中的紫色、蓝色和橙色分别表示该基因被≥3、2和1个hub转录因子调控"

图8
加权基因共表达网络中基因的GO(A)和KEGG(B)富集图"
表6
淮麦48籽粒不同发育时期枢纽TFs基因的表达量与蛋白质含量的相关性分析"
| 表型/基因<BOLD>P</BOLD>henotype/gene | 5 d | 10 d | 15 d | 20 d | 25 d | 30 d | 相关性系数r | |
|---|---|---|---|---|---|---|---|---|
| 蛋白质含量GPC (%) | 15.59 | 15.04 | 14.15 | 12.81 | 12.16 | 13.20 | ||
| TraesCS2A03G0362900 | 8.60 | 5.85 | 2.99 | 1.32 | 0.90 | 0.56 | 0.93** | |
| TraesCS4D03G0036300 | 13.19 | 7.96 | 2.72 | 2.36 | 1.57 | 2.39 | 0.89* | |
| TraesCS5A03G0079200 | 5.32 | 3.77 | 1.95 | 0.64 | 0.50 | 0.54 | 0.95** | |
| TraesCS5A03G0135400 | 21.54 | 48.33 | 35.01 | 15.00 | 11.12 | 8.03 | 0.67n.s. | |
| TraesCS5B03G0740100 | 136.13 | 83.19 | 35.66 | 28.30 | 19.17 | 15.06 | 0.89* | |
| TraesCS7D03G0590500 | 284.97 | 153.86 | 63.75 | 55.35 | 33.35 | 24.53 | 0.87* |
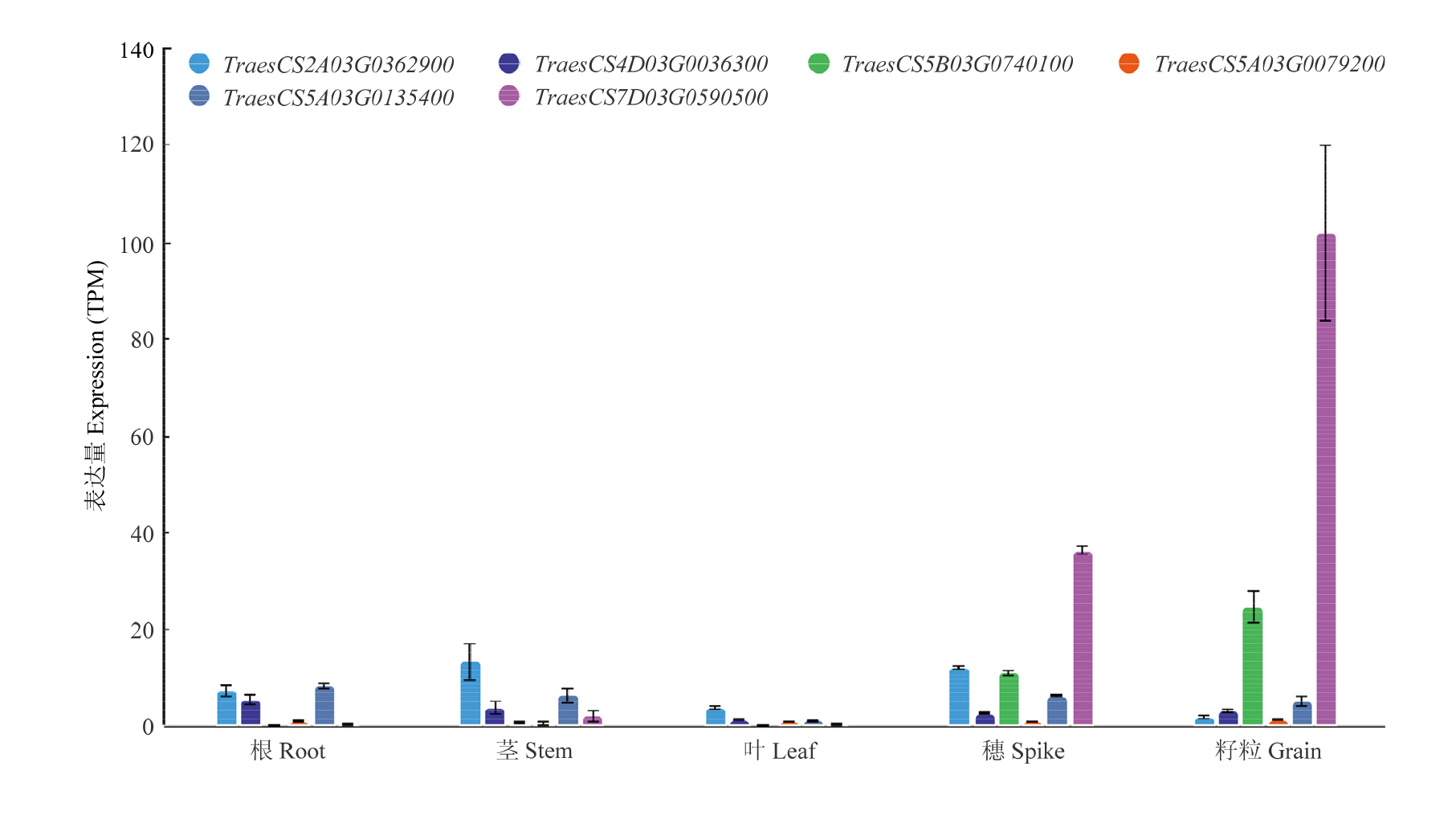
图9
WheatOmics中枢纽TFs基因在不同组织中的表达"
| [1] |
doi: 10.1186/1471-2164-9-121 pmid: 18325108 |
| [2] |
刘旭, 李立会, 黎裕, 谭光万, 周美亮. 作物及其种质资源与人文环境的协同演变学说. 植物遗传资源学报, 2022, 23(1): 1-11.
doi: 10.13430/j.cnki.jpgr.20211202001 |
|
doi: 10.13430/j.cnki.jpgr.20211202001 |
|
| [3] |
|
| [4] |
doi: 10.1002/pmic.200900792 pmid: 20641138 |
| [5] |
|
| [6] |
doi: 10.1093/plcell/koaa040 pmid: 33955492 |
| [7] |
|
| [8] |
doi: 10.1111/pbi.13524 pmid: 33305445 |
| [9] |
doi: 10.3389/fpls.2017.01133 pmid: 28702045 |
| [10] |
|
| [11] |
International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science, 2018, 361(6403): eaar7191.
|
| [12] |
|
| [13] |
doi: 10.1111/tpj.15538 pmid: 34634158 |
| [14] |
doi: 10.7150/ijbs.22619 pmid: 29483831 |
| [15] |
|
| [16] |
doi: 10.1186/1471-2105-9-559 pmid: 19114008 |
| [17] |
陈敏氡, 王彬, 刘建汀, 李永平, 白昌辉, 叶新如, 裘波音, 温庆放, 朱海生. 基于转录组和WGCNA筛选丝瓜果长和果径发育调控相关基因. 中国农业科学, 2023, 56(22): 4506-4522. doi: 10.3864/j.issn.0578-1752.2023.22.012.
|
|
|
|
| [18] |
曾健, 徐先超, 徐昱斐, 王秀成, 于海燕, 冯贝贝, 邢光南. 利用动态转录组学挖掘大豆百粒重候选基因. 作物学报, 2021, 47(11): 2121-2133.
doi: 10.3724/SP.J.1006.2021.04249 |
|
doi: 10.3724/SP.J.1006.2021.04249 |
|
| [19] |
杨闯, 王玲, 全成滔, 余良倩, 戴成, 郭亮, 傅廷栋, 马朝芝. 甘蓝型油菜盐胁迫响应基因表达谱分析及共表达网络的构建. 作物学报, 2024, 50(1): 237-250.
doi: 10.3724/SP.J.1006.2024.34076 |
|
|
|
| [20] |
doi: 10.1111/pbi.14107 pmid: 37589238 |
| [21] |
ISO/TS16634-2:2009. Food products - Determination of the total nitrogen content by combustion according to the Dumas principle and calculation of the crude protein content -Part2: Cereals, pulses and milled cereal products. Switzerland: International organization for standardization, Geneva: 2009: 25.
|
| [22] |
doi: 10.1111/pbi.13809 pmid: 35315196 |
| [23] |
doi: 10.1126/science.1133649 pmid: 17124321 |
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
pmid: 24828307 |
| [28] |
doi: 10.1105/tpc.18.00422 pmid: 30242039 |
| [29] |
|
| [30] |
doi: 10.1104/pp.106.082826 pmid: 16798940 |
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
doi: 10.1111/j.1365-313X.2009.04101.x pmid: 20003164 |
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
pmid: 16163610 |
| [39] |
|
| [40] |
|
| [1] | 曹海顺, 周东源, 王瑞, 施招婉, 吴廷全, 张长远. 弱光胁迫短下胚轴黄瓜种质鉴定及其遗传位点挖掘[J]. 中国农业科学, 2026, 59(6): 1286-1301. |
| [2] | 费耀莹, 王迪, 唐伟杰, 郭彩丽, 张小虎, 邱小雷, 程涛, 姚霞, 江冲亚, 朱艳, 曹卫星, 郑恒彪. 基于无人机多源影像融合的水稻籽粒蛋白质含量估测[J]. 中国农业科学, 2026, 59(1): 41-56. |
| [3] | 韦萍, 潘炬忠, 朱德平, 邵胜雪, 陈珊珊, 韦雅倩, 高维维. OsDREB1J调控水稻籽粒大小的功能研究[J]. 中国农业科学, 2025, 58(8): 1463-1478. |
| [4] | 王梦园, 魏倩睿, 李海艳, 杨巧敏, 宇军, 黄炜, 逯明辉. 辣椒MADS-box转录因子基因CaAGL61的耐热功能分析[J]. 中国农业科学, 2025, 58(8): 1604-1616. |
| [5] | 刁邓超, 李云丽, 孟祥宇, 季颂涵, 孙玉晨, 马学红, 李杰, 冯永佳, 李春莲, 吴建辉, 曾庆东, 韩德俊, $\boxed{\hbox{王长发}}$, 郑炜君. 小麦TaGRAS34-5A的克隆及耐热功能研究[J]. 中国农业科学, 2025, 58(4): 617-634. |
| [6] | 张林琳, 宫瑞, 崔彦玲, 钟雄辉, 李烨, 李然红, 潜宗伟. 利用VIGS分析SmWRKY30在茄子抗青枯病中的作用[J]. 中国农业科学, 2025, 58(3): 548-563. |
| [7] | 穆赢通, 路景诗, 张雨桐, 石凤翎. 基于转录组和WGCNA的直立型花苜蓿抗旱关键基因识别[J]. 中国农业科学, 2025, 58(21): 4528-4543. |
| [8] | 邹霈怡, 刘美艳, 王颖, 李然红. 狗枣猕猴桃AkNAC2的克隆及功能研究[J]. 中国农业科学, 2025, 58(19): 3985-3999. |
| [9] | 董雪, 陈梦秋, 邵晋, 吴学友, 唐培安. 基于WGCNA的稻谷储藏期间差异基因挖掘与品质调控网络构建[J]. 中国农业科学, 2025, 58(14): 2885-2903. |
| [10] | 阮桥君, 毛苗苗, 张媛媛, 林晓蓉, 陈忠正. 茶树‘英红9号’乙胺合成关键酶基因CsAlaDC转录因子的筛选[J]. 中国农业科学, 2025, 58(12): 2427-2438. |
| [11] | 姜兴林, 于连伟, 付涵, 艾妞, 崔荧钧, 李好海, 夏子豪, 袁虹霞, 李洪连, 杨雪, 施艳. 转录因子NbMYB1R1通过促进活性氧积累抑制病毒侵染[J]. 中国农业科学, 2024, 57(8): 1490-1505. |
| [12] | 朱俊杰, 张鑫悦, 潘梦影, 张静雯, 郑琦, 李玉玲, 董永彬. 玉米EIN3/EIL家族基因ZmEIL9调控籽粒发育[J]. 中国农业科学, 2024, 57(18): 3522-3532. |
| [13] | 陈晓涓, 王海菊, 王富敏, 雍清青, 黄顺满, 屈燕. 基于WGCNA鉴定全缘叶绿绒蒿类黄酮合成途径关键基因[J]. 中国农业科学, 2024, 57(15): 3053-3070. |
| [14] | 胡丹丹, 罗润琪, 梁瑞英, 汪磊, 梁琳, 司红彬, 丁家波, 汤新明. ApiAP2转录因子家族调控弓形虫生长发育的研究进展[J]. 中国农业科学, 2024, 57(13): 2687-2697. |
| [15] | 白斌, 张怀志, 杜久元, 张晓洋, 何瑞, 伍玲, 张哲, 张耀辉, 曹世勤, 刘志勇. 西北条锈菌源区冬小麦育种抗条锈病基因的利用现状与策略[J]. 中国农业科学, 2024, 57(1): 4-17. |
|
||