






中国农业科学 ›› 2020, Vol. 53 ›› Issue (11): 2207-2218.doi: 10.3864/j.issn.0578-1752.2020.11.007
秦艳红,王永江,王爽,乔奇,田雨婷,张德胜,张振臣( )
)
收稿日期:2019-11-07
接受日期:2019-11-28
出版日期:2020-06-01
发布日期:2020-06-09
通讯作者:
张振臣
作者简介:秦艳红,E-mail: qinyanhong6040@163.com。
基金资助:
QIN YanHong,WANG YongJiang,WANG Shuang,QIAO Qi,TIAN YuTing,ZHANG DeSheng,ZHANG ZhenChen()
Received:2019-11-07
Accepted:2019-11-28
Online:2020-06-01
Published:2020-06-09
Contact:
ZhenChen ZHANG
摘要:
【目的】对甘薯羽状斑驳病毒(Sweet potato feathery mottle virus,SPFMV)O株系中国分离物(SPFMV-O-Ch1)和RC株系中国分离物(SPFMV-RC-Ch1)的基因组全序列进行克隆,明确SPFMV-O-Ch1和SPFMV-RC-Ch1的基因组结构特征及其遗传变异情况,为研究甘薯羽状斑驳病毒的致病机制打下基础。【方法】根据GenBank中登录的SPFMV基因组全序列设计2对简并引物和3对特异性引物,利用RT-PCR方法,从感染SPFMV的甘薯叶片中扩增SPFMV O株系和RC株系中国分离物的基因组全长序列,将目的片段分别克隆到pMD19-T载体上,经序列测定、分析和拼接,获得SPFMV-O-Ch1和SPFMV-RC-Ch1的全序列,利用DNAMAN和MEGA7对SPFMV基因组全序列及不同编码区序列进行遗传变异和系统进化树分析,利用RDP软件分析SPFMV基因组重组情况。【结果】经序列测定和拼接,结果表明SPFMV-O-Ch1和SPFMV-RC-Ch1基因组分别包含10 922和10 851 nt,均包含一个开放阅读框,分别由10 557和10 482 nt组成,编码一个多聚蛋白,分别由3 518和3 493个氨基酸残基组成。两个分离物均在P1蛋白内编码一个P1N-PISPO蛋白,在P3蛋白内编码一个P3N-PIPO蛋白。基因组全序列核苷酸一致性分析表明,SPFMV-O-Ch1与SPFMV-RC-Ch1的一致性为87.3%,与GenBank登录的其他分离物基因组全序列一致性为86.0%—95.8%,与Ruk73分离物的一致性最高,为95.8%,与11-1分离物的一致性最低,为86.0%。SPFMV-RC-Ch1与GenBank登录的其他分离物基因组全序列一致性为85.9%—98.7%,与IS90分离物的一致性最高,为98.7%,与Aus1-2B分离物的一致性最低,为85.9%。基于多聚蛋白基因核苷酸序列的遗传进化树分析表明,SPFMV-O-Ch1与Ordinary、10-O和17-O等O株系的分离物形成一个分支,SPFMV-RC-Ch1与S、IS90和CW137等RC株系的分离物形成一个分支。重组分析结果表明,O-Ch1分离物中发现3个重组事件,分别发生在7 731—9 710、135—10 012和4 825—6 948 nt,RC-Ch1没有发现重组事件。【结论】我国的SPFMV-O-Ch1和SPFMV-RC-Ch1分离物的基因组结构与其他分离物相同,O-Ch1与O株系分离物一致性较高,RC-Ch1与RC株系分离物一致性较高,O-Ch1分离物检测到3个重组事件,RC-Ch1未发现重组事件。
秦艳红,王永江,王爽,乔奇,田雨婷,张德胜,张振臣. 甘薯羽状斑驳病毒O株系和RC株系中国分离物全基因组 序列分析及其遗传特征[J]. 中国农业科学, 2020, 53(11): 2207-2218.
QIN YanHong,WANG YongJiang,WANG Shuang,QIAO Qi,TIAN YuTing,ZHANG DeSheng,ZHANG ZhenChen. Complete Nucleotide Sequence Analysis and Genetic Characterization of the Sweet potato feathery mottle virus O and RC Strains Isolated from China[J]. Scientia Agricultura Sinica, 2020, 53(11): 2207-2218.
表1
本研究所用分离物的名称、株系、来源和登录号信息"
| 序号Number | 分离物名称Isolate name | 株系Strain | 地理来源Geographic origin | 登录号Accession number |
|---|---|---|---|---|
| 1 | RC-Ch1 | RC | 中国China | KY296451 |
| 2 | O-Ch1 | O | 中国China | KY296450 |
| 3 | CW137 | RC | 韩国South Korea | KP115608 |
| 4 | IS90 | RC | 韩国South Korea | KP115610 |
| 5 | GJ122 | O | 韩国South Korea | KP115609 |
| 6 | S | RC | 日本Japan | D86371 |
| 7 | 10-O | O | 日本Japan | AB439206 |
| 8 | Ordinary | O | 日本Japan | AB465608 |
| 9 | 17-O | O | 日本Japan | AB509454 |
| 10 | 11-8 | RC | 美国USA | MH782223 |
| 11 | 95-204R | RC | 美国USA | MH782224 |
| 12 | 11-1 | RC | 美国USA | MH778648 |
| 13 | TFSW-1J | RC | 美国USA | MH782227 |
| 14 | 95-2T | O | 美国USA | MH782225 |
| 15 | Aus7D | RC | 澳大利亚Australia | MF572046 |
| 16 | Aus12D | RC | 澳大利亚Australia | MF572049 |
| 17 | Aus8-8CA | RC | 澳大利亚Australia | MG656422 |
| 18 | Aus11D | RC | 澳大利亚Australia | MF572048 |
| 19 | Aus15-21CA | RC | 澳大利亚Australia | MG656429 |
| 20 | Aus11-13CA | RC | 澳大利亚Australia | MG656425 |
| 21 | Aus6-3B | RC | 澳大利亚Australia | MG656420 |
| 22 | Aus9-9B-2 | RC | 澳大利亚Australia | MG656423 |
| 23 | Aus10-10D | RC | 澳大利亚Australia | MG656424 |
| 24 | Aus13-18B-1 | RC | 澳大利亚Australia | MG656427 |
| 25 | Aus4D | RC | 澳大利亚Australia | MF572047 |
| 26 | Aus3D | RC | 澳大利亚Australia | MF572052 |
| 27 | Aus7-7B | RC | 澳大利亚Australia | MG656421 |
| 28 | Aus2D | RC | 澳大利亚Australia | MF572051 |
| 29 | Aus4-3A | RC | 澳大利亚Australia | MG656418 |
| 30 | Aus9D | RC | 澳大利亚Australia | MF572054 |
| 31 | Aus14-20CA | RC | 澳大利亚Australia | MG656428 |
| 32 | Aus12-16B | RC | 澳大利亚Australia | MG656426 |
| 33 | Aus16-24A | RC | 澳大利亚Australia | MG656430 |
| 34 | Aus3-2BC | RC | 澳大利亚Australia | MG656417 |
| 35 | Aus17-11D | O | 澳大利亚Australia | MG656431 |
| 36 | Aus5-3B | O | 澳大利亚Australia | MG656419 |
| 37 | Aus13B | O | 澳大利亚Australia | MF572050 |
| 38 | Aus1-2B | O | 澳大利亚Australia | MG656415 |
| 39 | Aus2-2BC | O | 澳大利亚Australia | MG656416 |
| 40 | UNB-01 | RC | 巴西Brazil | MF185715 |
| 41 | TM33C | O | 东帝汶East Timor | MF572055 |
| 42 | TM64B | O | 东帝汶East Timor | MF572053 |
| 43 | TM66B | O | 东帝汶East Timor | MF572056 |
| 44 | Ruk73 | O | 乌干达Uganda | KP729265 |
| 45 | RC-Arg | RC | 阿根廷Argentina | KF386014 |
| 46 | O-Arg | O | 阿根廷Argentina | KF386013 |
| 47 | SRF109a | O | 肯尼亚Kenya | MH264535 |
| 48 | ZA-O | O | 南非South Africa | KT069222 |
| 49 | Piu3 | EA | 秘鲁Peru | FJ155666 |
| 50 | AM-MB2 | 西班牙Spain | KU511268 |
表2
引物序列及目的片段大小"
| 扩增片段 Amplified fragment | 引物名称 Primer name | 引物序列 Primer sequence (5′→3′) | 目的片段大小 Expected size (bp) |
|---|---|---|---|
| O-1/RC-1 | 1-F | AATAACACAACWCAAYACAACAYAASAAAACT | 4784/4711 |
| 4711-R | GTGATATCATCTAARCTYTGATG | ||
| O-2 | 4689-F | CATCARAGYTTAGATGATATCAC | 5692 |
| 10380-R | CATATCGCGCAAGACTCATATC | ||
| O-3 | SPFMV-O-4160-F | AAGCGACAGAGCAACAGACTTATGTACTA | 1124 |
| SPFMV-O-5284-R | GATATTGCGTTGTAAATTCAACCTCACGTC | ||
| RC-2 | SPFMV-RC-4260-F | AAGTTTGTGAATATGCTACTAGTCATCA | 3814 |
| SPFMV-RC-8220-R | TACCTTCTAGGTCTGTGATCAGTTTAGAC | ||
| RC-3 | SPFMV-RC-7580-F | CCGATAGCTAGCGTCATATGCCATCTCG | 3426 |
| SPFMV-11000-R | TTGGCTCGATCACGAACCAAAAAGGCT |

图1
O-Ch1和RC-Ch1分离物基因组不同片段RT-PCR产物 M:DL5000分子量标准 DL5000 marker;1:O-1扩增片段Amplification of O-1;2:O-2扩增片段Amplification of O-2;3:O-3扩增片段Amplification of O-3;4:RC-1扩增片段Amplification of RC-1;5:RC-2扩增片段Amplification of RC-2;6:RC-3扩增片段Amplification of RC-3"

表3
O-Ch1、RC-Ch1与其他分离物核苷酸和氨基酸序列一致性"
| 基因组区域 Genome region | O-Ch1蛋白大小及与48个分离物比对 Protein size of O-Ch1 and comparison with 48 isolates | RC-Ch1蛋白大小及与48个分离物比对 Protein size of RC-Ch1 and comparison with 48 isolates | O-Ch1与RC-Ch1比对 Comparison of O-Ch1 and RC-Ch1 | ||||
|---|---|---|---|---|---|---|---|
| 蛋白大小 (氨基酸) Protein size (aa) | 核苷酸 一致性 nt identity (%) | 氨基酸 一致性 aa identity (%) | 蛋白大小 (氨基酸) Protein size (aa) | 核苷酸 一致性 nt identity (%) | 氨基酸 一致性 aa identity (%) | 核苷酸(氨基酸) 一致性 nt (aa) identity (%) | |
| 全长Complete sequence | - | 86.0-95.8 | - | - | 85.9-98.7 | - | 87.3 (-) |
| 多聚蛋白Polyprotein | 3518 | 87.0-95.8 | 92.1-97.6 | 3493 | 87.0-98.8 | 92.8-99.3 | 88.2 (94.0) |
| P1 | 689 | 83.4-97.8 | 81.2-96.4 | 664 | 82.8-99.0 | 80.6-99.2 | 85.4 (83.9) |
| HC | 458 | 83.3-98.2 | 91.9-98.9 | 458 | 83.4-99.0 | 92.1-100.0 | 84.0 (93.2) |
| P3 | 352 | 93.2-98.5 | 95.5-98.6 | 352 | 92.8-99.3 | 94.3-99.4 | 93.8 (96.0) |
| 6K1 | 52 | 92.9-100.0 | 92.3-100.0 | 52 | 91.7-98.7 | 94.2-100.0 | 94.2 (98.1) |
| CI | 643 | 89.0-98.0 | 97.0-99.7 | 643 | 89.7-98.9 | 97.5-99.8 | 89.9 (98.0) |
| 6K2 | 53 | 91.2-98.7 | 94.3-100.0 | 53 | 89.3-98.7 | 96.2-100.0 | 91.8 (98.1) |
| NIa-VPg | 192 | 78.1-97.7 | 90.6-100.0 | 192 | 83.9-99.3 | 89.6-100.0 | 84.4 (97.4) |
| NIa-Pro | 243 | 83.4-96.3 | 93.4-98.8 | 243 | 79.0-98.6 | 92.6-99.6 | 85.9 (96.3) |
| NIb | 521 | 86.3-97.3 | 93.1-98.5 | 521 | 85.3-98.7 | 92.9-99.8 | 89.5 (96.2) |
| CP | 315 | 76.7-96.6 | 94.0-99.7 | 315 | 79.9-98.9 | 95.6-99.7 | 92.5 (96.8) |
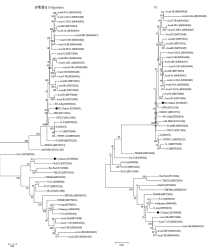
图2
SPFMV分离物多聚蛋白和P1蛋白核苷酸序列遗传进化分析"
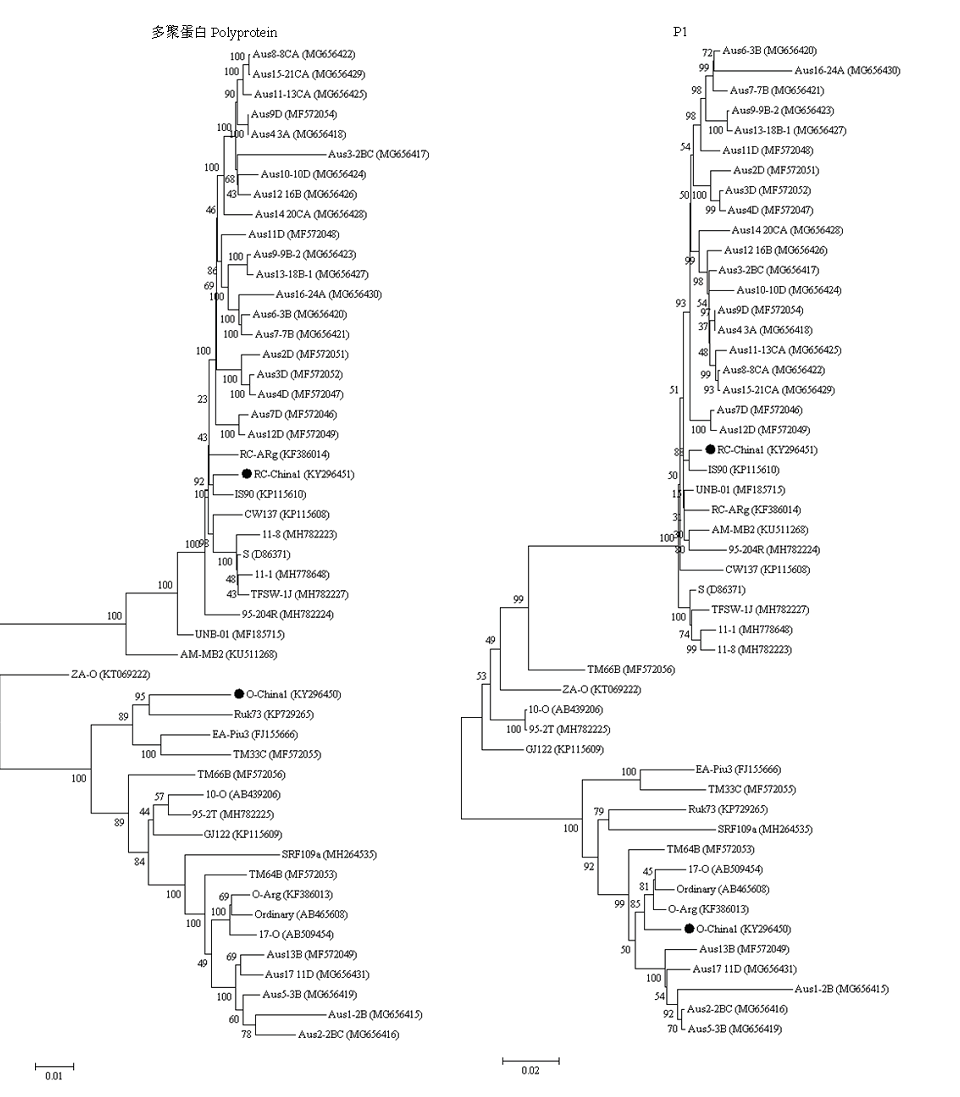
图3
SPFMV分离物CI蛋白和CP蛋白核苷酸序列遗传进化分析"

表4
O-Ch1分离物的重组分析"
| 序号 Number | 主要亲本 Major parent | 次要亲本 Minor parent | 位置 Position (nt) | P值P-Value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RDP | GENECONV | BootScan | MaxChi | Chimaera | SiScan | 3Seq | ||||
| 1 | Ordinary- AB465608 | TM33C- MF572055 | 7731- 9710 | 5.583×10-33 | 8.317×10-17 | 7.407×10-31 | 3.758×10-20 | 3.466×10-22 | 2.430×10-25 | 1.095×10-44 |
| 2 | EA-Piu3- FJ155666 | Aus5-3B- MG656419 | 135- 10012 | - | - | - | 8.783×10-4 | 7.437×10-3 | 1.803×10-20 | 1.561×10-56 |
| 3 | Aus13B- MF572050 | EA-Piu3- FJ155666 | 4825- 6948 | - | - | - | 1.104×10-3 | 1.071×10-2 | - | 1.256×10-2 |
| [1] |
RÄNNÄLI M, CZEKAJ V, JONES R A C, FLETCHER J D, DAVIS R I, MU L, VALKONEN J P T . Molecular characterization of Sweet potato feathery mottle virus (SPFMV) isolates from Easter Island, French Polynesia, New Zealand and southern Africa. Plant Disease, 2009,93(9):933-939.
doi: 10.1094/PDIS-93-9-0933 |
| [2] | 孟清, 张鹤龄, 张喜印, 杨永嘉, 邢继英, 宋伯符 . 甘薯羽状斑驳病毒的分离与提纯. 植物病理学报, 1994,24(3):227-232. |
| MENG Q, ZHANG H L, ZHANG X Y, YANG Y J, XING J Y, SONG B F . Isolation, purification of Sweet potato feathery mottle virus. Acta Phytopathologica Sinica, 1994,24(3):227-232. (in Chinese) | |
| [3] |
MUKASA S B, TAIRO F, KREUZE J F, KULLAYA A, RUBAIHAYO P R, VALKONEN J P T . Coat protein sequence analysis reveals occurrence of new strains of Sweet potato feathery mottle virus in Uganda and Tanzania. Virus Genes, 2003,27(1):49-56.
doi: 10.1023/A:1025172402230 |
| [4] | MOYER J W, KENNEDY G G . Purification and properties of Sweet potato feathery mottle virus. Phytopathology, 1978,68:998-1004. |
| [5] | KARYEINA R F, KREUZE J F, GIBSON R W, VALKONEN J P T . Synergistic interactions of a potyvirus and a phloem-limited crinivirus in sweet potato plants. Virology, 2000,269(1):26-36. |
| [6] | UNTIVEROS M, FUENTES S, KREUZE J . Molecular variability of Sweet potato feathery mottle virus and other potyviruses infecting sweet potato in Peru. Archive of Virology, 2008,153(3):473-483. |
| [7] | TAIRO F, MUKASA S B, JONES R A C, KULLAYA A, RUBAIHAYO P R, VALKONEN J P T, . Unravelling the genetic diversity of the three main viruses involved in sweet potato virus disease (SPVD), and its practical implications. Molecular Plant Pathology, 2005,6(2):199-211. |
| [8] | 李汝刚, 蔡少华, SALAZAR L F . 中国甘薯病毒的血清学检测. 植物病理学报, 1990,20(3):189-194. |
| LI R G, CAI S H, SALAZAR L F . Serological detection of viruses on sweet potato in China. Acta Phytopathologica Sinica, 1990,20(3):189-194. (in Chinese) | |
| [9] | 乔奇, 张振臣, 张德胜, 秦艳红, 田雨婷, 王永江 . 中国甘薯病毒种类的血清学和分子检测. 植物病理学报, 2012,42(1):10-16. |
| QIAO Q, ZHANG Z C, ZHANG D S, QIN Y H, TIAN Y T, WANG Y J . Serological and molecular detection of viruses infecting sweet potato in China. Acta Phytopathologica Sinica, 2012,42(1):10-16. (in Chinese) | |
| [10] |
QIN Y H, ZHANG Z C, QIAO Q, ZHANG D S, TIAN Y T, WANG Y J . Molecular variability of Sweet potato chlorotic stunt virus (SPCSV) and five potyviruses infecting sweet potato in China. Archive of Virology, 2013,158(2):491-495.
doi: 10.1007/s00705-012-1503-8 |
| [11] | 王晓华, 张振臣, 乔奇, 秦艳红, 张德胜, 田雨婷 . 甘薯羽状斑驳病毒外壳蛋白基因的分子变异. 植物保护, 2012,38(2):114-116. |
| WANG X H, ZHANG Z C, QIAO Q, QIN Y H, ZHANG D S, TIAN Y T . Molecular variation of Sweet potato feathery mottle virus coat protein gene. Plant Protection, 2012,38(2):114-116. (in Chinese) | |
| [12] | SAKAI J, MORI M, MORISHITA A, TANAKA M, HANADA K, USUGI T, NISHIGUCHI M . Complete nucleotide sequence and genome organization of Sweet potato feathery mottle virus (S strain) genomic RNA: The large coding region of the P1gene. Archive of Virology, 1997,142(8):1553-1562. |
| [13] |
KREUZE J F, PEREZ A, UNTIVEROS M, QUISPE D, FUENTES S, BARKER I, SIMON R . Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology, 2009,388(1):1-7.
doi: 10.1016/j.virol.2009.03.024 |
| [14] | YAMASAKI S, SAKAI J, FUJI S, KAMISOYAMA S, EMOTO K, OHSHIMA K, HANADA K . Comparisons among isolates of Sweet potato feathery mottle virus using complete genomic RNA sequences. Archive of Virology, 2010,155(5):795-800. |
| [15] | KWAK H R, KIM J, KIM M K, SEO J K, JUNG M N, KIM J S, LEE S C, CHOI H S . Molecular characterization of five potyviruses infecting Korean sweet potatoes based on analyses of complete genome sequences. The Plant Pathology Journal, 2015,31(4):388-401. |
| [16] |
MINGOT A, VALLI A, RODAMILANS B, LEON D S, BAULCOMBE D C, GARCIA J A, LOPEZ-MOYA J J . The P1N-PISPO trans-frame gene of sweet potato feathery mottle potyvirus is produced during virus infection and functions as RNA silencing suppressor. Journal of Virology, 2016,90(7):3543-3557.
doi: 10.1128/JVI.02360-15 |
| [17] |
MAINA S, BARBETTI M J, EDWARDS O, DE ALMEIDA L, XIMENES A, JONES R A C . Sweet potato feathery mottle virus and Sweet potato virus C from East Timorese and Australian sweetpotato: Biological and molecular properties, and biosecurity implications. Plant Disease, 2018,102(3):589-599.
doi: 10.1094/PDIS-08-17-1156-RE |
| [18] |
MAINA S, BARBETTI M J, MARTIN D P, EDWARDS O R, JONES R A C . New isolates of Sweet potato feathery mottle virus and Sweet potato virus C: Biological and molecular properties, and recombination analysis based on complete genomes. Plant Disease, 2018,102(10):1899-1914.
doi: 10.1094/PDIS-12-17-1972-RE |
| [19] |
CLARK C A, DAVIS J A, ABAD J A, CUELLAR W J, FUENTES S, KREUZE J F, GIBSON R W, MUKASA S B, TUGUME A K, TAIRO F D, VALKONEN J P T . Sweetpotato viruses: 15 years of progress on understanding and managing complex diseases. Plant Disease, 2012,96(2):168-185.
doi: 10.1094/PDIS-07-11-0550 |
| [20] | UNTIVEROS M, QUISPE D, KREUZE J . Analysis of complete genomic sequences of isolates of the Sweet potato feathery mottle virus strains C and EA: Molecular evidence for two distinct potyvirus species and two P1 protein domains. Archive of Virology, 2010,155(12):2059-2063. |
| [21] | LIU Q L, WANG Y J, ZHANG Z C, LV H, QIAO Q, QIN Y H, ZHANG D S, TIAN Y T, WANG S, LI J Q . Diversity of sweepoviruses infecting sweet potato in China. Plant Disease, 2017,101(12):2098-2103. |
| [22] | 张凤桐, 程林发, 耿超, 田延平, 原雪峰, 白艳菊, 李向东 . 一株PVY NTN-NW黑龙江马铃薯分离物的检测鉴定 . 植物病理学报, 2019,49(4):512-519. |
| ZHANG F T, CHENG L F, GENG C, TIAN Y P, YUAN X F, BAI Y J, LI X D . Detection and identification of a Potato virus Y (PVY) NTN-NW isolate from potato in Heilongjiang, China. Acta Phytopathologica Sinica, 2019,49(4):512-519. (in Chinese) | |
| [23] | 李科, 时洪伟, 荆陈沉, 孙现超, 周常勇, 青玲 . ACLSV山东苹果分离物基因组重组及CP序列多样性分析. 中国农业科学, 2015,48(14):2857-2867. |
| LI K, SHI H W, JING C C, SUN X C, ZHOU C Y, QING L . Analysis of genome recombination and CP sequence diversity of ACLSV apple isolate from Shandong. Scientia Agricultura Sinica, 2015,48(14):2857-2867. (in Chinese) | |
| [24] | 李汝刚, 朱笑梅, 薛爱红, 蔡少华, 王小凤 . 甘薯病毒病的研究. I甘薯羽状斑驳病毒的分离、鉴定. 植物病理学报, 1992,22(4):319-322. |
| LI R G, ZHU X M, XUE A H, CAI S H, WANG X F . Identification of Langfang isolate of Sweet potato feathery mottle virus. Acta Phytopathologica Sinica, 1992,22(4):319-322. (in Chinese) | |
| [25] | 朱作为, 薛启汉, 杨永嘉, 邢继英 . 甘薯羽状斑驳病毒的分离和提纯. 江苏农业学报, 1994,10(1):47-49. |
| ZHU Z W, XUE Q H, YANG Y J, XING J Y . Isolation and purification of Sweet potato feathery mottle virus. Jiangsu Journal of Agricultural Sciences, 1994,10(1):47-49. (in Chinese) | |
| [26] | 杨崇良, 路兴波, 王升吉, 尚佑芬, 赵玖华, 李长松 . 甘薯羽状斑驳病毒(SPFMV)生物学性状研究. 山东农业科学, 2001,33(1):26-29. |
| YANG C L, LU X B, WANG S J, SHANG Y F, ZHAO J H, LI C S . Biological characteristics research on Sweet potato feathery mottle virus. Shandong Agricultural Sciences, 2001,33(1):26-29. (in Chinese) | |
| [27] | 张振臣, 李大伟, 陈健夫, 于嘉林, 乔奇, 靳秀兰 . 甘薯羽状斑驳病毒外壳蛋白基因在大肠杆菌中的表达及特异抗血清的制备. 农业生物技术学报, 2000,8(2):177-179. |
| ZHANG Z C, LI D W, CHEN J F, YU J L, QIAO Q, JIN X L . Overexpression of Sweet potato feathery mottle virus coat protein in E. coli and preparation of its specific antiserum. Journal of Agricultural Biotechnology, 2000,8(2):177-179. (in Chinese) | |
| [28] | 张盼, 兰新芝, 乔奇, 张德胜, 秦艳红, 田雨婷, 王爽, 张振臣 . 甘薯病毒病害(SPVD)的多重RT-PCR检测方法及其应用. 植物保护, 2013,39(2):86-90. |
| ZHANG P, LAN X Z, QIAO Q, ZHANG D S, QIN Y H, TIAN Y T, WANG S, ZHANG Z C . Development and application of a multiplex RT-PCR detection method for sweet potato virus disease (SPVD). Plant Protection, 2013,39(2):86-90. (in Chinese) | |
| [29] | 许泳清, 李华伟, 刘中华, 邱永祥, 罗文彬, 纪荣昌, 汤浩, 邱思鑫, 余华 . 甘薯羽状斑驳病毒(SPFMV)ELISA鉴定及RT-PCR检测方法的建立. 福建农业学报, 2013,28(12):1267-1272. |
| XU Y Q, LI H W, LIU Z H, QIU Y X, LUO W B, JI R C, TANG H, QIU S X, YU H . ELISA identification and development of RT-PCR detection of Sweet potato feathery mottle virus. Fujian Journal of Agricultural Sciences, 2013,28(12):1267-1272. (in Chinese) | |
| [30] | 王丽, 王振东, 乔奇, 秦艳红, 张德胜, 田雨婷, 王爽, 张立军, 张振臣 . 甘薯羽状斑驳病毒实时荧光定量PCR检测方法的建立. 沈阳农业大学学报, 2013,44(2):129-135. |
| WANG L, WANG Z D, QIAO Q, QIN Y H, ZHANG D S, TIAN Y T, WANG S, ZHANG L J, ZHANG Z C . Development of real-time fluorescent quantitative PCR assay for detection of Sweet potato feathery mottle virus. Journal of Shenyang Agricultural University, 2013,44(2):129-135. (in Chinese) | |
| [31] | 何海旺, 何虎翼, 谭冠宁, 刘义明, 何新民, 唐洲萍, 李丽淑, 王晖 . 反向斑点杂交法快速检测甘薯羽状斑驳病毒和甘薯G病毒. 南方农业学报, 2014,45(1):43-48. |
| HE H W, HE H Y, TAN G N, LIU Y M, HE X M, TANG Z P, LI L S, WANG H . Detection of SPFMV and SPVG by using reverse dot blot hybridization system. Journal of Southern Agriculture, 2014,45(1):43-48. (in Chinese) | |
| [32] | 许泳清, 李华伟, 邱思鑫, 刘中华, 邱永祥, 罗文彬, 纪荣昌, 汤浩, 余华 . 甘薯羽状斑驳病毒和褪绿矮化病毒双重RT-PCR检测方法的建立. 福建农业学报, 2014,29(11):1114-1117. |
| XU Y Q, LI H W, QIU S X, LIU Z H, QIU Y X, LUO W B, JI R C, TANG H, YU H . Development of dutiplex RT-PCR for the detection of SPFMV and SPCSV in sweet potato. Fujian Journal of Agricultural Sciences, 2014,29(11):1114-1117. (in Chinese) | |
| [33] | 李华伟, 许泳清, 邱思鑫, 刘中华, 邱永祥, 罗文彬, 汤浩, 余华 . 侵染甘薯的SPCSV、SPVG、SPFMV多重RT-PCR检测方法的建立及应用. 核农学报, 2015,29(8):1464-1470. |
| LI H W, XU Y Q, QIU S X, LIU Z H, QIU Y X, LUO W B, TANG H, YU H . Establishment and application of a multiplex RT-PCR detection method for SPCSV, SPVG and SPFMV Infecting sweetpotato. Journal of Nuclear Agricultural Sciences, 2015,29(8):1464-1470. (in Chinese) | |
| [34] | 卢会翔, 吕长文, 吴正丹, 罗凯, 尹旺, 杨航, 王季春, 张凯 . 甘薯羽状斑驳病毒(SPFMV)和甘薯褪绿矮化病毒(SPCSV)荧光定量RT-PCR检测方法的建立. 中国农业科学, 2016,49(1):90-102. |
| LU H X, LÜ C W, WU Z D, LUO K, YIN W, YANG H, WANG J C, ZHANG K . Development of detection method for Sweet potato feathery mottle virus (SPFMV) and Sweet potato chlorotic stunt virus (SPCSV) through fluorescence quantitative RT-PCR. Scientia Agricultura Sinica, 2016,49(1):90-102. (in Chinese) | |
| [35] | 蒋素华, 程喜梅, 宋彩霞, 许申平, 梁芳, 崔波 . 三种甘薯病毒多重RT-PCR检测技术的建立. 植物保护, 2017,43(1):126-130. |
| JIANG S H, CHENG X M, SONG C X, XU S P, LIANG F, CUI B . Establishment of multiplex RT-PCR system for detection of three viruses in sweet potato. Plant Protection, 2017,43(1):126-130. (in Chinese) | |
| [36] | 姜姗姗, 冯佳, 张眉, 王升吉, 辛志梅, 吴斌, 辛相启 . 甘薯羽状斑驳病毒RT-LAMP快速检测方法的建立. 中国农业科学, 2018,51(7):1294-1302. |
| JIANG S S, FENG J, ZHANG M, WANG S J, XIN Z M, WU B, XIN X Q . Development of RT-LAMP assay for rapid detection of Sweet potato feathery mottle virus (SPFMV). Scientia Agricultura Sinica, 2018,51(7):1294-1302. (in Chinese) | |
| [37] | 黄广学, 孟利前, 朱建晨, 张进, 李庞博, 肖海峻 . 甘薯羽状斑驳病毒(SPFMV)和褪绿矮化病毒(SPCSV)的双重RT-PCR检测技术体系构建. 沈阳农业大学学报, 2018,49(6):724-729. |
| HUANG G X, MENG L Q, ZHU J C, ZHANG J, LI P B, XIAO H J . Establishment of duplex RT-PCR detection method for SPFMV and SPCSV infecting sweet potato. Journal of Shenyang Agricultural University, 2018,49(6):724-729. (in Chinese) | |
| [38] | 肖海峻, 孟利前, 朱建晨, 张进, 李庞博, 黄广学 . 甘薯羽状斑驳病毒的RT-PCR检测技术构建. 分子植物育种, 2019,17(13):4302-4306. |
| XIAO H J, MENG L Q, ZHU J C, ZHANG J, LI P B, HUANG G X . Construction of RT-PCR detection technique for Sweet potato feathery mottle virus. Molecular Plant Breeding, 2019,17(13):4302-4306. (in Chinese) | |
| [39] | 张振臣, 乔奇, 秦艳红, 张德胜, 田雨婷 . 我国发现由甘薯褪绿矮化病毒和甘薯羽状斑驳病毒协生共侵染引起的甘薯病毒病害. 植物病理学报, 2012,42(3):328-333. |
| ZHANG Z C, QIAO Q, QIN Y H, ZHANG D S, TIAN Y T . First evidence for occurrence of sweet potato virus disease (SPVD) caused by dual infection of Sweet potato feathery mottle virus and Sweet potato chlorotic stunt virus in China. Acta Phytopathologica Sinica, 2012,42(3):328-333. (in Chinese) | |
| [40] | 张新新, 王旭芳, 林坚淳, 余竟成, 黄立飞, 董章勇 . 甘薯毁灭性病毒病害(SPVD)的研究进展. 中国农学通报, 2019,35(1):118-126. |
| ZHANG X X, WANG X F, LIN J C, YU J C, HUANG L F, DONG Z Y . Sweetpotato virus diseases (SPVD): Research progress. Chinese Agricultural Science Bulletin, 2019,35(1):118-126. (in Chinese) | |
| [41] | KREUZE J F, KARYEIJA R F, GIBSON R W, VALKONEN J P T . Comparisons of coat protein gene sequences show that Ease African isolates of Sweet potato feathery mottle virus form a genetically distinct group. Archive of Virology, 2000,145(3):567-574. |
| [1] | 黄勋和,翁茁先,李威娜,王庆,何丹林,罗威,张细权,杜炳旺. 中国地方品种黄鸡线粒体DNA D-loop遗传多样性研究[J]. 中国农业科学, 2022, 55(22): 4526-4538. |
| [2] | 靳容,刘明,赵鹏,张强强,张爱君,唐忠厚. 甘薯丝裂原活化蛋白激酶MPK6对低温胁迫的响应[J]. 中国农业科学, 2021, 54(20): 4265-4273. |
| [3] | 张林林,智慧,汤沙,张仁梁,张伟,贾冠清,刁现民. 谷子抽穗时间基因SiTOC1的表达与单倍型变异分析[J]. 中国农业科学, 2021, 54(11): 2273-2286. |
| [4] | 武炳超, 童磊, 杜昭昌, 胡家菱, 张欢, 陈燚, 刘伟, 张新全, 黄琳凯. 60Co-γ射线对2种狼尾草属牧草的诱变效应[J]. 中国农业科学, 2019, 52(3): 414-427. |
| [5] | 王玉龙, 黄冰艳, 王思雨, 杜培, 齐飞艳, 房元瑾, 孙子淇, 郑峥, 董文召, 张新友. 四倍体野生种花生A.monticola全基因组SSR的开发与特征分析[J]. 中国农业科学, 2019, 52(15): 2567-2580. |
| [6] | 姜珊珊,冯佳,张眉,王升吉,辛志梅,吴斌,辛相启. 甘薯羽状斑驳病毒RT-LAMP快速检测方法的建立[J]. 中国农业科学, 2018, 51(7): 1294-1302. |
| [7] | 王亚飞,阮涛,周彦,王雪峰,吴根土,孙现超,周常勇,青玲. 甜橙和柚中CTV强弱毒株系p20的遗传变异[J]. 中国农业科学, 2017, 50(7): 1343-1350. |
| [8] | 严静,江雨,樊秀彩,姜建福,张颖,孙海生,刘崇怀. 中国11种野生葡萄果皮中黄烷-3-醇类物质的组成及含量[J]. 中国农业科学, 2017, 50(5): 890-902. |
| [9] | 王琳,李新凤,徐玉梅,畅引东,王建明. 山西省植物病原镰孢菌种群分布及遗传变异分析[J]. 中国农业科学, 2017, 50(10): 1802-1816. |
| [10] | 赵培方,赵俊,刘家勇,昝逢刚,夏红明,P.A. Jackson, J. Basnayake, N.G. Inman-Bamber, 杨昆,赵丽萍,覃伟,陈学宽,赵兴东,范源洪. 干旱胁迫对甘蔗4个生理指标遗传变异的影响[J]. 中国农业科学, 2017, 50(1): 28-37. |
| [11] | 沈林林,邹文超,高芳銮,詹家绥. 采用多基因联合方法鉴定福建长乐和福清产区马铃薯Y病毒株系组成[J]. 中国农业科学, 2016, 49(20): 3918-3926. |
| [12] | 卢会翔,吕长文,吴正丹,罗凯,尹旺,杨航,王季春,张凯. 甘薯羽状斑驳病毒(SPFMV)和甘薯褪绿矮化病毒(SPCSV)荧光定量RT-PCR检测方法的建立[J]. 中国农业科学, 2016, 49(1): 90-102. |
| [13] | 耿立英,张传生,赵书雨,陈娟,巩元芳,刘铮铸,朱文进,李祥龙. 鸡mir-1658前体基因多态性分析[J]. 中国农业科学, 2015, 48(19): 3919-3930. |
| [14] | 张芮,张宗营,高利平,冀晓昊,毛志泉,许海峰,王楠,吴树敬,陈学森. 苹果绵肉与脆肉株系果实质地差异的分子机理[J]. 中国农业科学, 2015, 48(18): 3676-3688. |
| [15] | 刘志斋,吴迅,李永祥,丁晓雨,王凤格,石云素,宋燕春,赵久然,黎裕,王天宇. 中国玉米地方品种种族的遗传变异评估[J]. 中国农业科学, 2015, 48(16): 3101-3111. |
|
||