






中国农业科学 ›› 2024, Vol. 57 ›› Issue (7): 1394-1406.doi: 10.3864/j.issn.0578-1752.2024.07.014
赵真坚( ), 王凯, 陈栋, 申琦, 余杨, 崔晟頔, 王俊戈, 陈子旸, 禹世欣, 陈佳苗, 王翔枫, 唐国庆()
), 王凯, 陈栋, 申琦, 余杨, 崔晟頔, 王俊戈, 陈子旸, 禹世欣, 陈佳苗, 王翔枫, 唐国庆()
收稿日期:2023-10-18
接受日期:2023-12-31
出版日期:2024-04-01
发布日期:2024-04-09
通信作者:
联系方式:
赵真坚,E-mail:530773281@qq.com。
基金资助:
ZHAO ZhenJian(), WANG Kai, CHEN Dong, SHEN Qi, YU Yang, CUI ShengDi, WANG JunGe, CHEN ZiYang, YU ShiXin, CHEN JiaMiao, WANG XiangFeng, TANG GuoQing()
Received:2023-10-18
Accepted:2023-12-31
Published:2024-04-01
Online:2024-04-09
摘要:
【背景】猪的肉质性状是重要的经济性状。研究影响各类肉质性状的分子机制,发掘关键基因,从而指导猪的遗传改良,对改善猪肉品质具有重要意义。当前肉质相关的机制研究主要是基于DNA的基因组研究,而对肉质性状的DNA甲基化研究以及结合基因组和甲基化组的综合分析却鲜有报道。【目的】通过基因组和DNA甲基化组联合分析,筛选、鉴定了影响猪肉质的潜在关键基因。为猪肉品质的遗传改良研究提供借鉴。【方法】检测了140头大白猪背最长肌的28个肉质性状,通过表观基因组关联分析(EWAS)与全基因组关联分析(GWAS)筛选了各性状显著关联的CpG和SNP位点。随后以SNP为协变量对GWAS和EWAS重叠的显著关联位点进行条件关联分析,进一步筛选具有独立效应的CpG位点。然后以CpG位点的甲基化水平为因变量,以SNP为自变量进行关联分析从而鉴定甲基化数量性状位点(meQTL)。最后使用顺式甲基化数量性状位点(cis-meQTL)作为工具变量进行孟德尔随机化分析,从而推断cis-meQTL与表型之间的因果关系,同时对位点进行注释,鉴定潜在的关键基因。【结果】(1)在屠宰45 min黄度值(b45min)、滴水损失(DL)、二十二碳六烯酸(C22:6n-3)3个肉质性状上,EWAS和GWAS在相同基因组区域鉴定到显著关联位点。(2)b45min的7个CpG位点在条件关联分析后仍保持显著,DL有1个CpG位点在条件关联分析后仍保持显著,而C22:6n-3的3个位点在使用SNP作为协变量分析后不再显著,表明EWAS鉴定的b45min的7个CpG位点和DL的1个CpG位点的显著关联不受附近的显著SNP影响。(3)b45min的7个CpG位点和DL的1个CpG位点共鉴定了10个meQTL,但绝大多数是trans-meQTL,只有一个CpG位点(SSC12:44 254 675 bp)鉴定到一个cis-meQTL,表明该位点可能受到近距离SNP调控。(4)孟德尔随机化分析显示该CpG位点(SSC12:44 254 675 bp)与b45min表型存在一定的因果关联。(5)对该位点注释发现,距离CpG位点(SSC12:44 254 675 bp)和其cis-meQTL最近的基因是NOS2,且CpG位点位于NOS2基因内。【结论】综合DNA甲基化组合基因组数据联合分析结果,可以推测NOS2基因是肉色性状关键候选基因,其DNA甲基化、SNP共同作用调控基因表达,进而影响肉色性状相关基因表达。
赵真坚, 王凯, 陈栋, 申琦, 余杨, 崔晟頔, 王俊戈, 陈子旸, 禹世欣, 陈佳苗, 王翔枫, 唐国庆. 基因组和DNA甲基化组联合分析筛选猪肉质性状关键基因[J]. 中国农业科学, 2024, 57(7): 1394-1406.
ZHAO ZhenJian, WANG Kai, CHEN Dong, SHEN Qi, YU Yang, CUI ShengDi, WANG JunGe, CHEN ZiYang, YU ShiXin, CHEN JiaMiao, WANG XiangFeng, TANG GuoQing. Integrated Aanalysis of Genome and DNA Methylation for Screening Key Genes Related to Pork Quality Traits[J]. Scientia Agricultura Sinica, 2024, 57(7): 1394-1406.
表1
GWAS和EWAS重叠的显著关联信号统计"
| 性状 Trait | GWAS关联 GWAS associations | EWAS关联 EWAS associations | ||
|---|---|---|---|---|
| 染色体SSC | 位点或区域 Position or Region (bp) | SNP数量 Number of SNP | CpG位点 CpG site (bp) | |
| b45min | 1 | 4049232 | 1 | SSC1:4667485 |
| b45min | 6 | 5191168-5483926 | 10 | SSC6:4737404 |
| b45min | 7 | 9770008-10877899 | 20 | SSC7:10489621 |
| b45min | 10 | 35692369 | 1 | SSC10:36329538 |
| b45min | 12 | 44218560 | 1 | SSC12:44254675 |
| b45min | 14 | 17744509-18820911 | 19 | SSC14:17859513 |
| b45min | 18 | 49231979-50010893 | 7 | SSC18:48429425 |
| b45min | 18 | 49231979-50010893 | 7 | SSC18:50979305 |
| DL | 7 | 121791317 | 1 | SSC7:120954182 |
| C22:6n-3 | 4 | 101860069-103765886 | 146 | SSC4:102796889 |
| C22:6n-3 | 4 | 101860069-103765886 | 146 | SSC4:103218200 |
| C22:6n-3 | 4 | 101860069-103765886 | 146 | SSC4:103552805 |
表2
对GWAS与EWAS共同的显著信号条件关联分析结果"
| 性状Trait | CpG位点 CpG site (bp) | 染色体SSC | 位置 Position (bp) | P value (EWAS) | P value (after correct) |
|---|---|---|---|---|---|
| b45min | SSC1: 4667485 | 1 | 4667485 | 3.95E-9 | 3.95E-9 |
| SSC6: 4737404 | 6 | 4737404 | 7.59E-9 | 0.35 | |
| SSC7: 10489621 | 7 | 10489621 | 1.53E-9 | 1.53E-9 | |
| SSC10: 36329538 | 10 | 36329538 | 2.76E-13 | 2.76E-13 | |
| SSC12: 44254675 | 12 | 44254675 | 5.45E-9 | 5.45E-9 | |
| SSC14: 17859513 | 14 | 17859513 | 1.26E-9 | 1.26E-9 | |
| SSC18: 48429425 | 18 | 48429425 | 1.06E-10 | 1.06E-10 | |
| SSC18: 50979305 | 18 | 50979305 | 1.18E-9 | 1.18E-9 | |
| DL | SSC7: 120954182 | 7 | 120954182 | 7.79E-9 | 7.79E-9 |
| C22:6n-3 | SSC4: 102796889 | 4 | 102796889 | 1.32E-12 | 0.99 |
| SSC4: 103218200 | 4 | 103218200 | 3.32E-9 | 0.13 | |
| SSC4: 103552805 | 4 | 103552805 | 1.32E-12 | 0.99 |
表3
甲基化数量性状位点鉴定结果"
| CpG位点 CpG site (bp) | 甲基化数量性状位点 meQTL | 顺式meQTL cis-meQTL | 反式meQTL trans-meQTL | SNP |
|---|---|---|---|---|
| SSC1:4667485 | 0 | 0 | 0 | 0 |
| SSC7:10489621 | 2 | 0 | 2 | 9 |
| SSC10:36329538 | 0 | 0 | 0 | 0 |
| SSC12:44254675 | 2 | 1 | 1 | 11 |
| SSC14:17859513 | 2 | 0 | 2 | 46 |
| SSC18:48429425 | 0 | 0 | 0 | 0 |
| SSC18:50979305 | 3 | 0 | 3 | 345 |
| SSC7:120954182 | 1 | 0 | 1 | 4 |
图1
CpG位点与基因组SNP关联分析的曼哈顿图"
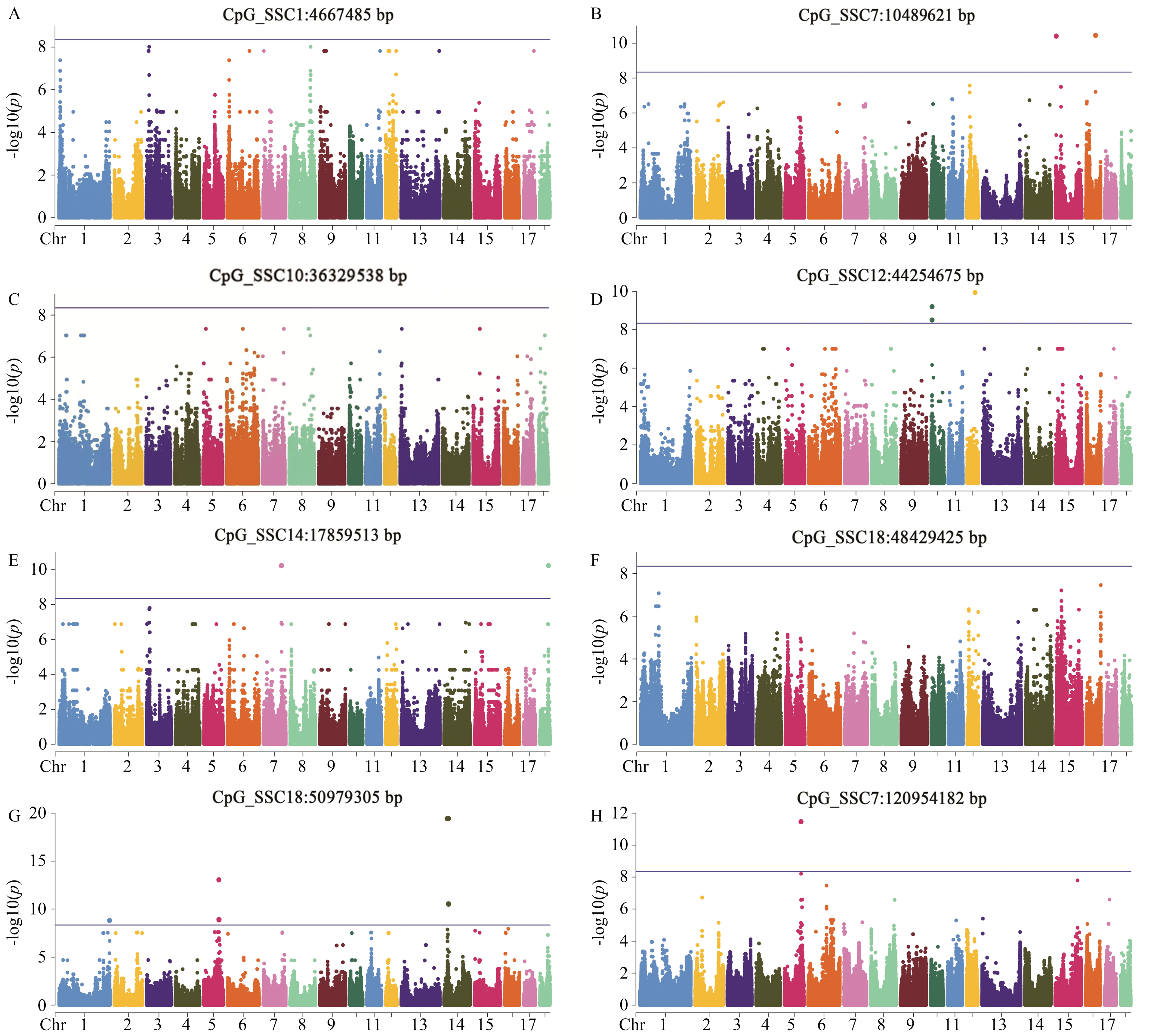
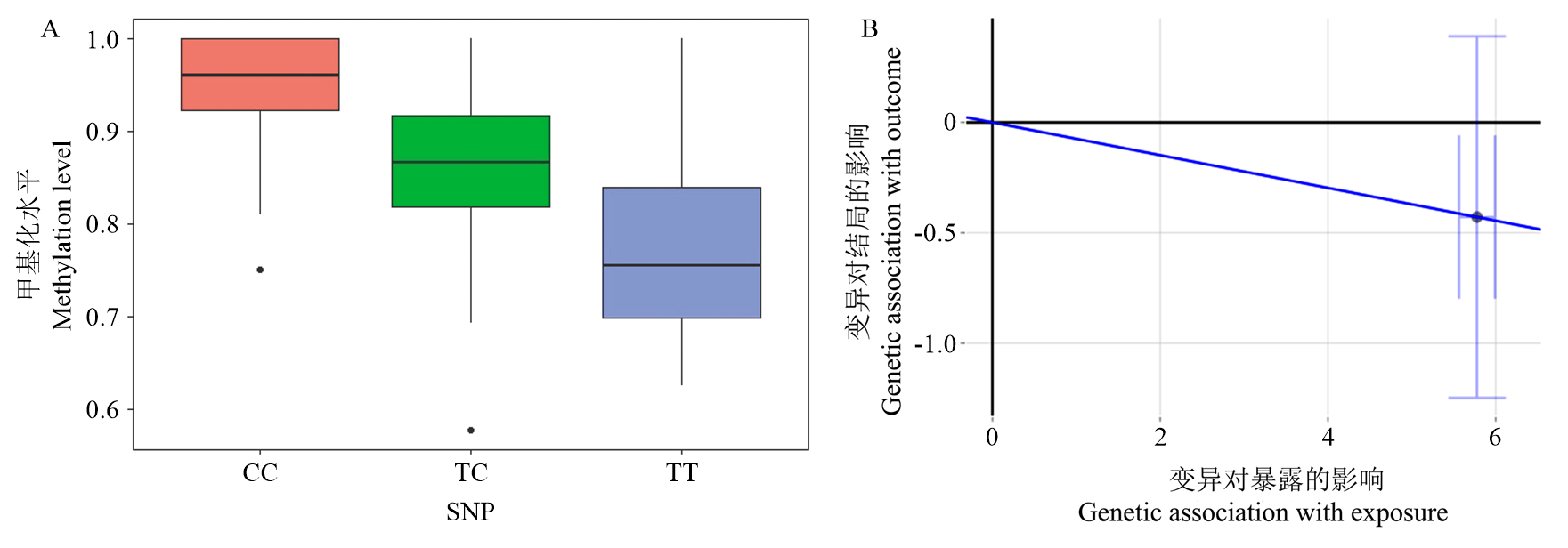
图2
cis-meQTL的SNP位点甲基化和孟德尔随机化分析 A. CpG位点SSC12:44 254 675 bp的cis-meQTL的箱线图,横坐标表示SNP基因型,纵坐标表示CpG甲基化水平;B. cis-meQTL的孟德尔随机化分析的散点图,横坐标表示SNP位点(SSC12:43 262 418 bp)对CpG 位点(SSC12:44 254 675 bp)甲基化的影响,纵坐标表示SNP位点(SSC12: 43 262 418 bp)对表型性状(b45min)的影响"
| [1] |
孙建广, 张石蕊. 猪肉品质研究进展. 中国猪业, 2008, 3(7): 58-59.
|
|
|
|
| [2] |
宋志芳, 解幼志, 刑荷岩, 芦春莲, 曹洪战. 影响猪肉质性状相关基因的研究进展. 中国猪业, 2017, 12(7): 48-50.
|
|
|
|
| [3] |
doi: 10.2527/jas.2006-114 pmid: 17202397 |
| [4] |
doi: 10.1080/10495398.2016.1243550 pmid: 27854153 |
| [5] |
李梦云, 郑萍, 李婉涛, 陈代文. PRKAG3基因在不同品种猪不同生长阶段骨骼肌中的表达差异及其表达量与肉质的关系. 动物营养学报, 2017, 29(5): 1661-1669.
|
|
|
|
| [6] |
doi: 10.1016/j.physbeh.2008.04.020 |
| [7] |
于太永. 脂肪细胞因子leptin和TNF: α对猪骨骼肌成肌细胞增殖分化的影响及其机制[D]. 杨凌: 西北农林科技大学, 2007.
|
|
|
|
| [8] |
李志娟. 猪心脏型脂肪酸结合蛋白研究进展. 养猪, 2021(2): 55-56.
|
|
|
|
| [9] |
doi: 10.1007/s11033-016-3969-z |
| [10] |
doi: 10.1016/j.meatsci.2019.05.013 |
| [11] |
pmid: 17488358 |
| [12] |
|
| [13] |
doi: 10.1016/j.meatsci.2012.03.018 |
| [14] |
周李生, 杨杰, 刘先先, 张志燕, 杨斌, 麻骏武. 苏太猪宰后72 h pH和肉色性状的全基因组关联分析. 中国农业科学, 2014, 47(3): 564-573. doi: 10.3864/j.issn.0578-1752.2014.03.016.
|
|
|
|
| [15] |
doi: 10.3389/fgene.2021.614087 |
| [16] |
doi: 10.3389/fgene.2021.748070 |
| [17] |
doi: 10.1093/nar/gkaa1203 pmid: 33434283 |
| [18] |
doi: 10.1007/s13353-020-00604-1 pmid: 33400132 |
| [19] |
doi: 10.3389/fphys.2021.697121 |
| [20] |
|
| [21] |
doi: 10.1111/jpn.v106.2 |
| [22] |
唐韶青, 张沅, 徐青, 孙东晓, 俞英. 不同动物部分组织基因组甲基化程度的差异分析. 农业生物技术学报, 2006, 14(4): 507-510.
|
|
|
|
| [23] |
doi: 10.1038/ncomms1854 pmid: 22617290 |
| [24] |
张小丽. 猪背部浅层和背部深层脂肪组织全基因组甲基化研究[D]. 雅安: 四川农业大学, 2013.
|
|
|
|
| [25] |
|
| [26] |
李娜. 基于GWAS的猪肉品质性状候选基因研究[D]. 北京: 中国农业大学, 2016.
|
|
|
|
| [27] |
秦一禾. 猪肌内脂肪含量的全基因组关联研究[D]. 昆明: 云南大学, 2017.
|
|
|
|
| [28] |
张俊杰. 利用多品种大样本群体精细定位影响猪背最长肌脂肪酸组成的QTL[D]. 南昌: 江西农业大学, 2018.
|
|
|
|
| [29] |
doi: 10.1093/bioinformatics/btu170 pmid: 24695404 |
| [30] |
doi: 10.1093/bioinformatics/btr167 pmid: 21493656 |
| [31] |
doi: 10.1038/nmeth.1923 pmid: 22388286 |
| [32] |
doi: 10.1093/bioinformatics/btp324 pmid: 19451168 |
| [33] |
doi: 10.1093/bioinformatics/btp352 pmid: 19505943 |
| [34] |
doi: 10.1038/ng.806 pmid: 21478889 |
| [35] |
doi: 10.1186/s13742-015-0047-8 pmid: 25722852 |
| [36] |
doi: 10.1038/ng.2310 pmid: 22706312 |
| [37] |
doi: 10.1093/bioinformatics/btm108 pmid: 17384015 |
| [38] |
doi: 10.1080/1828051X.2023.2247003 |
| [39] |
|
| [40] |
doi: 10.3389/fgene.2020.00060 pmid: 32180791 |
| [41] |
doi: 10.1093/ije/dyt093 pmid: 24062299 |
| [42] |
doi: 10.4161/epi.25501 pmid: 23811543 |
| [43] |
|
| [44] |
doi: 10.1038/nrg.2017.32 pmid: 28555657 |
| [45] |
|
| [46] |
doi: 10.2217/epi.15.88 pmid: 26639554 |
| [47] |
doi: 10.1093/ije/dyz190 pmid: 31549173 |
| [48] |
doi: 10.1038/nature20784 |
| [49] |
doi: 10.4161/15592294.2014.969637 pmid: 25424692 |
| [50] |
doi: 10.1038/srep43261 pmid: 28256596 |
| [51] |
|
| [52] |
doi: 10.1017/S0016672305007597 |
| [53] |
pmid: 7511942 |
| [54] |
doi: 10.1016/j.ejphar.2003.09.034 |
| [55] |
|
| [56] |
doi: 10.1021/ic400697a pmid: 23768169 |
| [57] |
pmid: 10491373 |
| [58] |
doi: 10.1042/BJ20090716 |
| [1] | 张颖, 石婷瑞, 曹瑞, 潘文秋, 宋卫宁, 王利, 聂小军. ICARDA引进-小麦苗期抗旱性的全基因组关联分析[J]. 中国农业科学, 2024, 57(9): 1658-1673. |
| [2] | 高晨曦, 郝陆洋, 胡悦, 李永祥, 张登峰, 李春辉, 宋燕春, 石云素, 王天宇, 黎裕, 刘旭洋. 干旱条件下玉米转座子插入关联的表观调控分析[J]. 中国农业科学, 2024, 57(6): 1034-1048. |
| [3] | 郭军, 邵丹, 窦套存, 马猛, 卢建, 胡玉萍, 王星果, 王强, 李永峰, 郭伟, 童海兵, 曲亮. 鸡产蛋期剩余采食量的随机回归分析及遗传标记筛选[J]. 中国农业科学, 2024, 57(22): 4568-4577. |
| [4] | 郭军, 曲亮, 邵丹, 马猛, 窦套存, 卢建, 胡玉萍, 王星果, 王强, 李永峰, 郭伟, 童海兵. 基于一步法全基因组关联分析解析蛋黄比率遗传结构[J]. 中国农业科学, 2024, 57(21): 4356-4366. |
| [5] | 白冰楠, 乔丹, 葛群, 栾玉娟, 刘小芳, 卢全伟, 牛皓, 龚举武, 巩万奎, ELAMEER ELSAMMAN, 闫浩亮, 李俊文, 刘爱英, 石玉真, 王海泽, 袁有禄. 陆地棉棉籽相关性状的QTN挖掘及候选基因筛选[J]. 中国农业科学, 2024, 57(15): 2901-2913. |
| [6] | 桂翠林, 马亮, 王银莹, 谢富贵, 赵彩宏, 王文淼, 李鑫, 王青, 高夕全. 玉米镰孢菌复合病原茎腐病抗性种质资源鉴定及抗性基因挖掘[J]. 中国农业科学, 2024, 57(13): 2509-2524. |
| [7] | 寿鑫月, 刘智, 陈玥含, 李晨辉, 孙宾成, 孙如建, 韩德志, 鹿文成, 申永辉, 王晓波, 闫龙. 冀北坝上大豆结瘤相关性状全基因组关联分析[J]. 中国农业科学, 2024, 57(11): 2102-2113. |
| [8] | 张玉梅, 丁文涛, 蓝新隆, 李清华, 胡润芳, 郭娜, 林国强, 赵晋铭. 大豆地方种质资源鲜籽粒可溶性糖含量的全基因组关联分析[J]. 中国农业科学, 2024, 57(11): 2079-2091. |
| [9] | 骆娜, 安炳星, 魏立民, 文杰, 赵桂苹. 全基因组关联分析筛选文昌鸡体尺性状相关分子标记[J]. 中国农业科学, 2024, 57(10): 2046-2060. |
| [10] | 李盛有, 王昌陵, 闫春娟, 张立军, 孙旭刚, 曹永强, 王文斌, 宋书宏. 大豆种质抗旱性评价及抗旱候选基因的挖掘[J]. 中国农业科学, 2024, 57(10): 1857-1869. |
| [11] | 任志强, 王晨阳, 寇忠云, 蔡瑞, 杨公社, 庞卫军. 运用计算机断层扫描技术活体评估种公猪瘦肉率、脂肪率和肌内脂肪含量[J]. 中国农业科学, 2023, 56(9): 1787-1799. |
| [12] | 谭力治, 赵毅强. 全基因组关联分析中混合模型的原理、优化与应用[J]. 中国农业科学, 2023, 56(9): 1617-1632. |
| [13] | 王慧玲, 闫爱玲, 王晓玥, 刘振华, 任建成, 徐海英, 孙磊. 葡萄果粒质量相关性状全基因组关联分析[J]. 中国农业科学, 2023, 56(8): 1561-1573. |
| [14] | 李慧, 张雨峰, 李晓刚, 王中华, 蔺经, 常有宏. 全基因组DNA甲基化和转录组联合分析鉴定杜梨耐盐相关转录因子[J]. 中国农业科学, 2023, 56(7): 1377-1390. |
| [15] | 杨明路, 张海亮, 罗汉鹏, 黄锡霞, 张翰林, 章施施, 王炎, 刘林, 郭刚, 王雅春. 基于智能项圈系统荷斯坦牛发情相关指标的遗传参数估计及全基因组关联分析[J]. 中国农业科学, 2023, 56(5): 995-1006. |
|
||