






中国农业科学 ›› 2022, Vol. 55 ›› Issue (20): 4075-4090.doi: 10.3864/j.issn.0578-1752.2022.20.017
• 畜牧·兽医 • 上一篇
王艳文( ),王梦静,张虹,高鑫鑫,郭晶(),李旭勇()
),王梦静,张虹,高鑫鑫,郭晶(),李旭勇()
收稿日期:2021-08-06
接受日期:2022-04-07
出版日期:2022-10-16
发布日期:2022-10-24
联系方式:
王艳文,E-mail: 15290068938@163.com。
基金资助:
WANG YanWen(),WANG MengJing,ZHANG Hong,GAO XinXin,GUO Jing(),LI XuYong()
Received:2021-08-06
Accepted:2022-04-07
Published:2022-10-16
Online:2022-10-24
摘要:
【目的】 通过分析1998—2021年间我国人感染H9N2亚型禽流感病例的发病时间、所在省份、年龄和性别等信息,明确H9N2亚型禽流感病毒的流行病学特征;通过分析人源H9N2亚型禽流感病毒的基因特征,阐明人源H9N2亚型禽流感病毒的遗传演化规律;为H9N2亚型禽流感病毒跨种间传播的预警和防控提供数据支撑。【方法】 基于流感基因数据库、病例报道和文献资料,获得1998—2021年我国人感染H9N2亚型禽流感病毒的病例信息和毒株序列数据。从时间、空间、性别和年龄的分布对感染病例进行分析,明确人源H9N2亚型禽流感病毒感染的流行病学特征。通过DNASTAR中的MegAlign软件对人源H9N2病毒的各基因片段的核苷酸序列进行同源性分析,利用MEGA7.0软件构建系统进化树和分析病毒蛋白关键位点,揭示遗传演化趋势和病毒蛋白关键氨基酸位点的变异情况。通过GISAID网站下载2019—2021年间我国H9N2亚型禽流感病毒的核苷酸序列,利用mafft比对后在MEGA7.0中查看人源与禽源H9N2病毒关键氨基酸位点的突变差异,揭示当前人源和禽源H9N2病毒可能引起的风险。【结果】 1998—2021年我国人感染H9N2亚型禽流感病毒病例共71例,从空间分布分析,病例分布于16个省市,其中91.55%的病例来自于南方12个省市;从时间分布分析,2013年以后,我国报道的感染病例呈增长趋势,2013—2021年累计感染病例数占总病例数的61.97%;从性别和年龄分布分析,男、女性别比为 1﹕1.68,感染病例主要见于幼儿和少儿,占总病例的74.14%。对人源H9N2病毒进行基因组比对分析,发现这些病毒均属于欧亚分支,但是这些病毒各基因片段的核苷酸序列同源性差异较大,HA、NA、PB2、PB1、PA、NP、M和NS的同源性分别为75.3%—100%、80.1%—100%、78.7%—100%、82.5%—100%、72.6%—100%、74.1%—100%、65.5%—100%、82.0%—100%;22株具有完整基因片段的病毒分为8个基因型,2003、2008和2013年的基因型与1999年的基因型有明显差异。1998—2021年共有42株人源H9N2病毒株上传HA序列,其中有38株病毒的HA蛋白发生Q226L的突变;共有30株人源H9N2病毒株上传PB2序列,其中9株病毒的PB2蛋白发生E627V突变,1株病毒的PB2蛋白发生E627K突变;1株病毒的PB2蛋白的701位点发生D701N突变,共有31株人源H9N2病毒株上传 NS与M序列,NS1蛋白的42位点均为S,M1蛋白的30和215位点的氨基酸分别为D和A。2019-2021年人源H9N2病毒的HA蛋白183与190位点、NS1蛋白42位点均发生突变,人源与禽源H9N2病毒的PB2蛋白701位点均未发生突变。【结论】 自2013年以来,我国人感染H9N2亚型禽流感病例数量呈增长趋势,且具有显著的地域、年龄和性别差异。1998年至今,人源H9N2病毒的基因同源性差异较大,不同分支间病毒基因重排频繁,形成了复杂的基因型,提示H9N2亚型禽流感病毒在不断地进化。人源H9N2病毒的关键氨基酸位点出现突变,且在2019—2021年人源比禽源H9N2病毒的关键位点突变率高,提示H9N2亚型禽流感病毒的跨种感染人的潜力逐渐增强。该结果丰富了对人源H9N2病毒认知,为H9N2亚型禽流感病毒防控提供参考。
王艳文,王梦静,张虹,高鑫鑫,郭晶,李旭勇. 1998-2021年我国人感染H9N2亚型禽流感病毒的遗传演化规律[J]. 中国农业科学, 2022, 55(20): 4075-4090.
WANG YanWen,WANG MengJing,ZHANG Hong,GAO XinXin,GUO Jing,LI XuYong. Evolution of Human H9N2 Avian Influenza Virus in China from 1998 to 2021[J]. Scientia Agricultura Sinica, 2022, 55(20): 4075-4090.
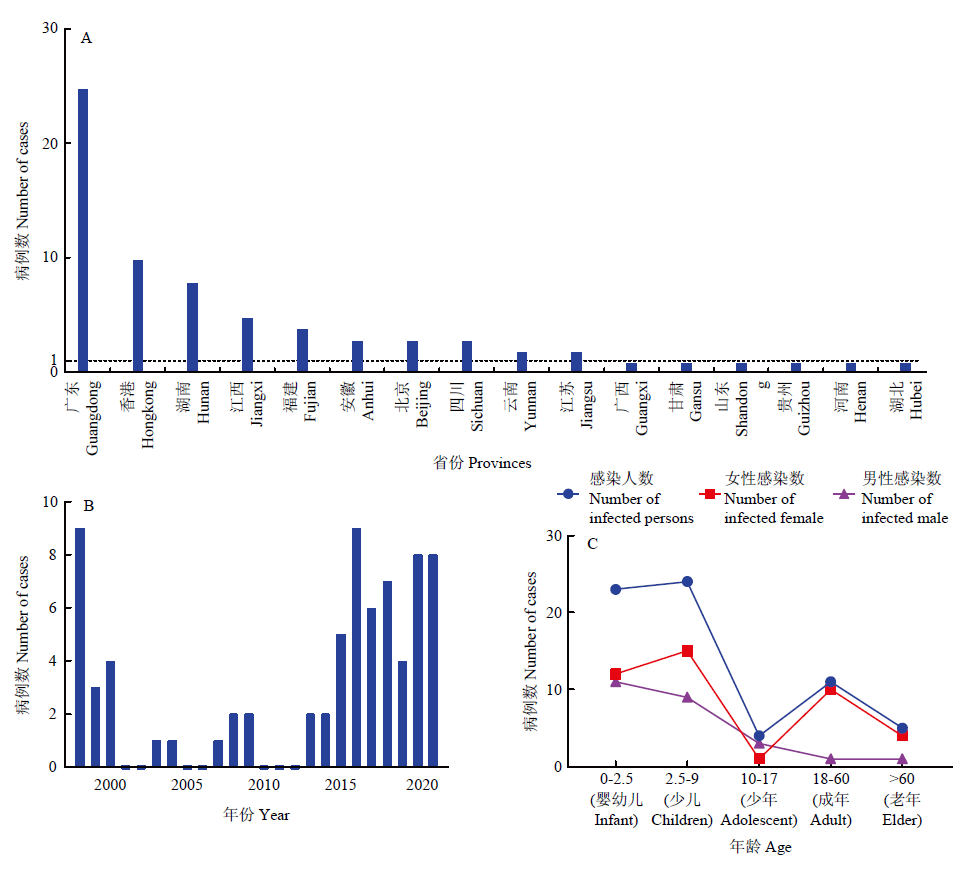
图1
1998—2021年全国人感染H9N2亚型AIV病例的分布图 A:全国人感染H9N2亚型AIV病例的空间分布;B:全国人感染H9N2亚型AIV病例的时间分布图;C:全国人感染H9N2亚型AIV病例的年龄和性别分布图"

图2
HA和NA基因进化树 A:HA基因进化树;B:NA基因进化树"

图3
内部基因进化树 A:PB2基因进化树;B:PB1基因进化树;C:PA基因进化树;D:NP基因进化树;E:M基因进化树;F:NS基因进化树"
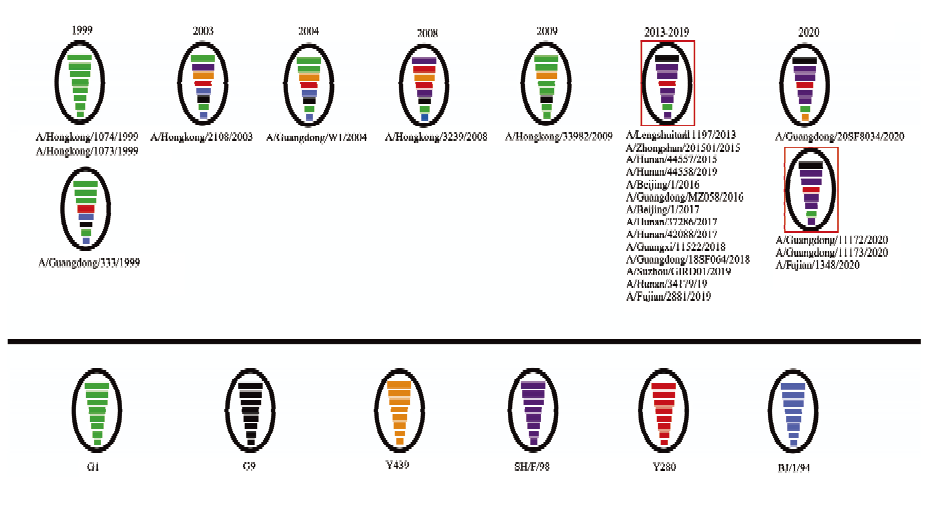
图4
1998-2021年感染人的H9N2亚型禽流感基因型演变"
表1
部分基因的关键位点分析"
| 人源H9N2分离株 Human H9N2 virus | HA | PB2 | NS1 | M1 | ||
|---|---|---|---|---|---|---|
| 226 | 627 | 701 | 42 | 30 | 215 | |
| A/Shantou/239/1998(H9N2) | Q | - | - | - | - | - |
| A/Shaoguan/408/1998(H9N2) | L | - | - | - | - | - |
| A/Shaoguan/447/1998(H9N2) | L | - | - | - | - | - |
| A/Guangzhou/333/1999(H9N2) | M | E | D | S | D | A |
| A/Hongkong/1073/1999(H9N2) | L | E | D | S | D | A |
| A/Hongkong/1074/1999(H9N2) | L | E | D | S | D | A |
| A/Nanchang/CH2/2000(H9N2) | L | - | - | - | - | - |
| A/Nanchang/CH3/2000(H9N2) | L | - | - | - | - | - |
| A/Nanchang/D1/2000(H9N2) | L | - | - | - | - | - |
| A/Nanchang/D2/2000(H9N2) | L | - | - | - | - | - |
| A/Hongkong/2108/2003(H9N2) | L | E | D | S | D | A |
| A/Guangdong/W1/2004(H9N2) | L | E | D | S | D | A |
| A/Hongkong/3239/2008(H9N2) | L | E | D | S | D | A |
| A/Hongkong/69955/2008(H9N2) | L | - | - | - | - | - |
| A/Hongkong/33982/2009(H9N2) | Q | E | N | S | D | A |
| A/Hongkong/35820/2009(H9N2) | Q | - | - | S | D | A |
| A/Lengshuitan/11197/2013(H9N2) | L | E | D | S | D | A |
| A/Guangdong/167/2014(H9N2) | - | E | D | S | D | A |
| A/Guangdong/79/2015(H9N2) | - | E | D | S | D | A |
| A/Hunan/44557/2015(H9N2) | L | E | D | S | D | A |
| A/Hunan/44558/2015(H9N2) | L | E | D | S | D | A |
| A/Zhongshan/201501/2015(H9N2) | L | E | D | S | D | A |
| A/Beijing/1/2016(H9N2) | L | V | D | S | D | A |
| A/Guangdong/MZ058/2016(H9N2) | L | K | D | S | D | A |
| A/Beijing/1/2017(H9N2) | L | V | D | S | D | A |
| A/Hunan/37286/2017(H9N2) | L | V | D | S | D | A |
| A/Hunan/42088/2017(H9N2) | L | V | D | S | D | A |
| A/Hunan/34179/2018(H9N2) | L | V | D | S | D | A |
| A/Guangxi/11522/2018(H9N2) | L | E | D | S | D | A |
| A/Anhui/39/2018(H9N2) | L | V | D | S | D | A |
| A/Guangdong/18SF003/2018(H9N2) | L | E | D | S | D | A |
| A/Guangdong/18SF064/2018(H9N2) | L | E | D | S | D | A |
| A/Fujian/2881/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/1187/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/1317/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/1423/2019(H9N2) | L | E | D | S | D | A |
| A/Hubei/1534/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/1565/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/1631/2019(H9N2) | L | - | - | - | - | - |
| A/Hubei/1693/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/1803/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/2081/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/2280/2019(H9N2) | L | E | D | S | D | A |
| A/Hubei/2299/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/2370/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/239/2019(H9N2) | L | E | D | S | D | A |
| A/Hubei/2462/2019(H9N2) | L | E | D | S | D | A |
| A/Hubei/2542/2019(H9N2) | L | E | D | S | D | A |
| A/Hubei/295/2019(H9N2) | L | V | D | S | D | A |
| A/Hubei/362/2019(H9N2) | L | E | D | S | D | A |
| A/Suzhou/GIRD01/2019(H9N2) | L | E | D | S | D | A |
| A/Fujian/1348/2020(H9N2) | L | E | D | S | D | A |
| A/Guangdong/20SF8034/2020(H9N2) | L | E | D | S | - | - |
| A/Guangdong/SF1634/2020(H9N2) | L | - | - | - | - | - |
| A/Guangdong/20SF15010/2020(H9N2) | L | - | - | - | - | - |
| A/Guangdong/11172/2020(H9N2) | L | E | D | S | D | A |
| A/Guizhou/13392/2020(H9N2) | L | - | - | - | - | - |
| A/Hunan/11173/2020(H9N2) | L | V | D | S | D | A |
| A/Shandong/01/2020(H9N2) | L | V | D | S | D | A |
| A/Sichuan/1619/2020(H9N2) | L | - | - | - | - | - |
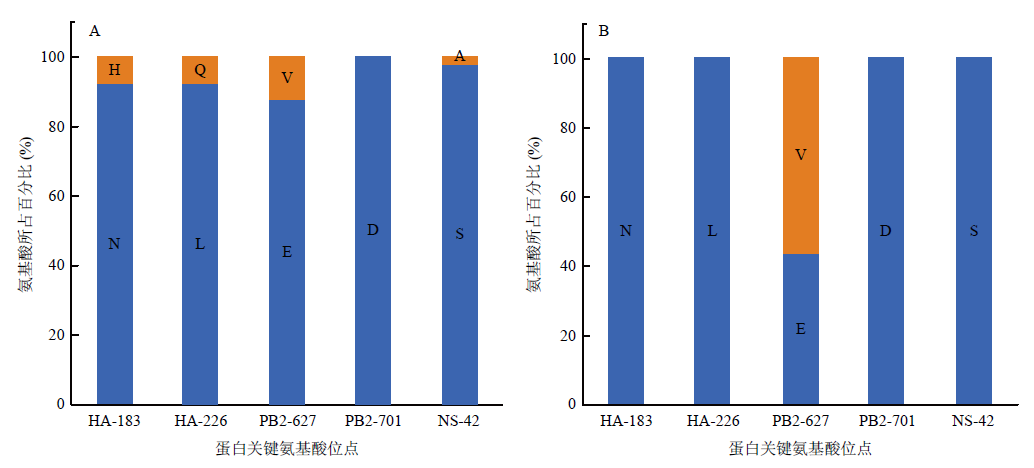
图5
2019-2021年H9N2分离株关键氨基酸位点分布 A:2019-2021年中国禽源H9N2病毒关键位点的分布;B:2019-2021年中国人源H9N2病毒关键位点的分布"
| [1] |
HOMME P J, EASTERDAY B C. Avian influenza virus infections. I. Characteristics of influenza A-Turkey-Wisconsin-1966 virus. Avian Diseases, 1970, 14(1): 66-74.
pmid: 4314007 |
| [2] |
ZHONG L, WANG X Q, LI Q H, LIU D, CHEN H Z, ZHAO M J, GU X B, HE L, LIU X W, GU M, PENG D X, LIU X F. Molecular mechanism of the airborne transmissibility of H9N2 avian influenza A viruses in chickens. Journal of Virology, 2014, 88(17): 9568-9578. doi: 10.1128/JVI.00943-14.
doi: 10.1128/JVI.00943-14 pmid: 24920791 |
| [3] |
ZHANG P H, TANG Y H, LIU X W, PENG D X, LIU W B, LIU H Q, LU S, LIU X F. Characterization of H9N2 influenza viruses isolated from vaccinated flocks in an integrated broiler chicken operation in Eastern China during a 5 year period (1998-2002). The Journal of General Virology, 2008, 89(Pt12): 3102-3112. doi: 10.1099/vir.0.2008/005652-0.
doi: 10.1099/vir.0. 2008/005652-0 |
| [4] |
SUN Y P, LIU J H. H9N2 influenza virus in China: a cause of concern. Protein & Cell, 2015, 6(1): 18-25. doi: 10.1007/s13238-014-0111-7.
doi: 10.1007/s13238-014-0111-7 |
| [5] |
PEACOCK T H P, JAMES J, SEALY J E, IQBAL M. A global perspective on H9N2 avian influenza virus. Viruses, 2019, 11(7): 620. doi: 10.3390/v11070620.
doi: 10.3390/v11070620 |
| [6] |
HE J, LIU B Y, GONG L, CHEN Z, CHEN X L, HOU S, YU J L, WU J B, XIA Z C, LATIF A, GAO R, SU B, LIU Y. Genetic characterization of the first detected human case of avian influenza A (H5N6) in Anhui Province, East China. Scientific Reports, 2018, 8: 15282. doi: 10.1038/s41598-018-33356-4.
doi: 10.1038/s41598-018-33356-4 pmid: 30327485 |
| [7] |
ZHANG Q Y, SHI J Z, DENG G H, GUO J, ZENG X Y, HE X J, KONG H H, GU C Y, LI X Y, LIU J X, WANG G J, CHEN Y, LIU L L, LIANG L B, LI Y Y, FAN J, WANG J L, LI W H, GUAN L Z, LI Q M, YANG H L, CHEN P C, JIANG L, GUAN Y T, XIN X G, JIANG Y P, TIAN G B, WANG X R, QIAO C L, LI C J, BU Z G, CHEN H L. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science, 2013, 341(6144): 410-414. doi: 10.1126/science.1240532.
doi: 10.1126/science.1240532 pmid: 23868922 |
| [8] |
LI X Y, CUI P F, ZENG X Y, JIANG Y P, LI Y B, YANG J X, PAN Y D, GAO X X, ZHAO C H, WANG J H, WANG K, DENG G H, GUO J. Characterization of avian influenza H5N3 reassortants isolated from migratory waterfowl and domestic ducks in China from 2015 to 2018. Transboundary and Emerging Diseases, 2019, 66(6): 2605-2610. doi: 10.1111/tbed.13324.
doi: 10.1111/tbed.13324 pmid: 31402584 |
| [9] |
CHEN H Y, YUAN H, GAO R B, ZHANG J X, WANG D Y, XIONG Y, FAN G Y, YANG F, LI X D, ZHOU J F, ZOU S M, YANG L, CHEN T, DONG L B, BO H, ZHAO X, ZHANG Y, LAN Y, SHU Y L. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. The Lancet, 2014, 383(9918):714-721. doi:10.1016/S0140-6736(14) 60111-2.
doi: 10.1016/S0140-6736(14)60111-2 |
| [10] |
YU X F, JIN T, CUI Y J, PU X Y, LI J, XU J, LIU G, JIA H J, LIU D, SONG S L, YU Y, XIE L, HUANG R J, DING H, KOU Y, ZHOU Y Y, WANG Y Y, XU X, YIN Y, WANG J, GUO C Y, YANG X W, HU L P, WU X P, WANG H L, LIU J, ZHAO G Q, ZHOU J Y, PAN J C, GAO G F, YANG R F, WANG J. Influenza H7N9 and H9N2 viruses: coexistence in poultry linked to human H7N9 infection and genome characteristics. Journal of Virology, 2014, 88(6): 3423-3431. doi: 10.1128/JVI.02059-13.
doi: 10.1128/JVI.02059-13 pmid: 24403589 |
| [11] |
KANDEIL A, EL-SHESHENY R, MAATOUQ A M, MOATASIM Y, SHEHATA M M, BAGATO O, RUBRUM A, SHANMUGANATHAM K, WEBBY R J, ALI M A, KAYALI G. Genetic and antigenic evolution of H9N2 avian influenza viruses circulating in Egypt between 2011 and 2013. Archives of Virology, 2014, 159(11): 2861-2876. doi: 10.1007/s00705-014-2118-z.
doi: 10.1007/s00705-014-2118-z pmid: 24990416 |
| [12] |
LIN Y P, SHAW M, GREGORY V, CAMERON K, LIM W, KLIMOV A, SUBBARAO K, GUAN Y, KRAUSS S, SHORTRIDGE K, WEBSTER R, COX N, HAY A. Avian-to-human transmission of H9N2 subtype influenza A viruses: relationship between H9N2 and H5N1 human isolates. Proceedings of the National Academy of Sciences of the United States of America, 2000, 97(17): 9654-9658. doi: 10.1073/pnas.160270697.
doi: 10.1073/pnas.160270697 pmid: 10920197 |
| [13] |
LV J, WEI B Z, YANG Y, YAO M L, CAI Y M, GAO Y W, XIA X Z, ZHAO X N, LIU Z H, LI X X, WANG H, YANG H L, ROESLER U, MIAO Z M, CHAI T J. Experimental transmission in Guinea pigs of H9N2 avian influenza viruses from indoor air of chicken houses. Virus Research, 2012, 170(1/2): 102-108. doi: 10.1016/j.virusres.2012.09.003.
doi: 10.1016/j.virusres.2012. 09.003 |
| [14] |
ZHANG K, ZHANG Z W, YU Z J, LI L, CHENG K H, WANG T C, HUANG G, YANG S T, ZHAO Y K, FENG N, FU J, QIN C, GAO Y W, XIA X Z. Domestic cats and dogs are susceptible to H9N2 avian influenza virus. Virus Research, 2013, 175(1): 52-57. doi: 10.1016/j.virusres.2013.04.004.
doi: 10.1016/j.virusres.2013.04.004 pmid: 23603563 |
| [15] |
ZHU Y C, ZHANG B, SUN Z H, WANG X J, FAN X H, GAO L X, LIANG Y, CHEN X Y, ZHANG Z F. Replication and pathology of duck influenza virus subtype H9N2 in chukar. Biomedical and Environmental Sciences, 2018, 31(4): 306-310. doi: 10.3967/bes2018.039.
doi: 10.3967/bes2018.039 pmid: 29773094 |
| [16] |
ALI M, YAQUB T, MUKHTAR N, IMRAN M, GHAFOOR A, SHAHID M F, NAEEM M, IQBAL M, SMITH G J D, SU Y C F. Avian influenza A(H9N2) virus in poultry worker, Pakistan, 2015. Emerging Infectious Diseases, 2019, 25(1): 136-139. doi: 10.3201/eid2501.180618.
doi: 10.3201/eid2501.180618 pmid: 30561309 |
| [17] |
NAGY A, METTENLEITER T C, ABDELWHAB E M. A brief summary of the epidemiology and genetic relatedness of avian influenza H9N2 virus in birds and mammals in the Middle East and North Africa. Epidemiology and Infection, 2017, 145(16): 3320-3333. doi: 10.1017/S0950268817002576.
doi: 10.1017/S0950268817002576 pmid: 29168447 |
| [18] |
孙莹, 张兵, 李岭, 黄小洁, 侯力丹, 刘丹, 李启红, 李俊平, 王乐元, 李慧姣, 杨承槐. 表达H9亚型禽流感病毒HA基因重组鸭肠炎病毒的构建. 中国农业科学, 2019, 52(23): 4398-4405. doi: 10.3864/j.issn.0578-1752.2019.23.020.
doi: 10. 3864/j.issn.0578-1752.2019.23.020 |
|
SUN Y, ZHANG B, LI L, HUANG X J, HOU L D, LIU D, LI Q H, LI J P, WANG L Y, LI H J, YANG C H. Construction of a recombinant duck enteritis virus expressing hemagglutinin of H9N2 avian influenza virus. Scientia Agricultura Sinica, 2019, 52(23): 4398-4405. doi: 10.3864/j.issn.0578-1752.2019.23.020. (in Chinese)
doi: 10. 3864/j.issn.0578-1752.2019.23.020 |
|
| [19] | 丁洁, 高玉伟, 桑晓宇, 程凯慧, 于志君, 张坤, 柴洪亮, 王铁成, 夏咸柱, 华育平. H9N2亚型AIV鼠肺适应株的获得及其氨基酸变异分析. 中国农业科学, 2015, 48(15): 3056-3063. |
| DING J, GAO Y W, SANG X Y, CHENG K H, YU Z J, ZHANG K, CHAI H L, WANG T C, XIA X Z, HUA Y P. The adaptation of H9N2 subtype AIV in mouse and analysis of amino acid mutation. Scientia Agricultura Sinica, 2015, 48(15): 3056-3063. (in Chinese) | |
| [20] |
LU J H, LIU X F, SHAO W X, LIU Y L, WEI D P, LIU H Q. Phylogenetic analysis of eight genes of H9N2 subtype influenza virus: a mainland China strain possessing early isolates' genes that have been circulating. Virus Genes, 2005, 31(2): 163-169. doi: 10.1007/s11262-005-1790-1.
doi: 10.1007/s11262- 005-1790-1 |
| [21] |
FUSARO A, MONNE I, SALVIATO A, VALASTRO V, SCHIVO A, AMARIN N M, GONZALEZ C, ISMAIL M M, AL-ANKARI A R, AL-BLOWI M H, KHAN O A, MAKEN ALI A S, HEDAYATI A, GARCIA GARCIA J, ZIAY G M, SHOUSHTARI A, AL QAHTANI K N, CAPUA I, HOLMES E C, CATTOLI G. Phylogeography and evolutionary history of reassortant H9N2 viruses with potential human health implications. Journal of Virology, 2011, 85(16): 8413-8421. doi: 10.1128/JVI.00219-11.
doi: 10.1128/JVI.00219-11 pmid: 21680519 |
| [22] |
XU K M, SMITH G J D, BAHL J, DUAN L, TAI H, VIJAYKRISHNA D, WANG J, ZHANG J X, LI K S, FAN X H, WEBSTER R G, CHEN H, PEIRIS J S M, GUAN Y. The genesis and evolution of H9N2 influenza viruses in poultry from Southern China, 2000 to 2005. Journal of Virology, 2007, 81(19): 10389-10401. doi: 10.1128/JVI.00979-07.
doi: 10.1128/JVI.00979-07 pmid: 17652402 |
| [23] |
SUN X J, BELSER J A, MAINES T R. Adaptation of H9N2 influenza viruses to mammalian hosts: a review of molecular markers. Viruses, 2020, 12(5): 541. doi: 10.3390/v12050541.
doi: 10.3390/v12050541 |
| [24] |
SORRELL E M, WAN H Q, ARAYA Y, SONG H C, PEREZ D R. Minimal molecular constraints for respiratory droplet transmission of an avian-human H9N2 influenza A virus. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106(18): 7565-7570. doi: 10.1073/pnas.0900877106.
doi: 10.1073/pnas.0900877106 pmid: 19380727 |
| [25] |
WAN H Q, PEREZ D R. Amino acid 226 in the hemagglutinin of H9N2 influenza viruses determines cell tropism and replication in human airway epithelial cells. Journal of Virology, 2007, 81(10): 5181-5191. doi: 10.1128/JVI.02827-06.
doi: 10.1128/JVI.02827-06 pmid: 17344280 |
| [26] |
GABRIEL G, HERWIG A, KLENK H D. Interaction of polymerase subunit PB2 and NP with importin alpha1 is a determinant of host range of influenza A virus. PLoS Pathogens, 2008, 4(2): e11. doi: 10.1371/journal.ppat.0040011.
doi: 10. 1371/journal.ppat.0040011 |
| [27] |
ZHANG H, LI X Y, GUO J, LI L, CHANG C, LI Y Y, BIAN C, XU K, CHEN H L, SUN B. The PB2 E627K mutation contributes to the high polymerase activity and enhanced replication of H7N9 influenza virus. The Journal of General Virology, 2014, 95(Pt 4): 779-786. doi: 10.1099/vir.0.061721-0.
doi: 10. 1099/vir.0.061721-0 |
| [28] |
GU M, XU L J, WANG X Q, LIU X F. Current situation of H9N2 subtype avian influenza in China. Veterinary Research, 2017, 48(1): 49. doi: 10.1186/s13567-017-0453-2.
doi: 10.1186/s13567-017-0453-2 pmid: 28915920 |
| [29] |
LI C, WANG S G, BING G X, CARTER R A, WANG Z J, WANG J L, WANG C X, WANG L, WU G, WEBSTER R G, WANG Y Q, SUN H L, SUN Y P, LIU J H, PU J. Genetic evolution of influenza H9N2 viruses isolated from various hosts in China from 1994 to 2013. Emerging Microbes & Infections, 2017, 6(1): 1-11. doi: 10.1038/emi.2017.94.
doi: 10.1038/emi. 2017.94 |
| [30] |
LI X Y, LIU B T, MA S J, CUI P F, LIU W Q, LI Y B, GUO J, CHEN H L. High frequency of reassortment after co-infection of chickens with the H4N6 and H9N2 influenza A viruses and the biological characteristics of the reassortants. Veterinary Microbiology, 2018, 222: 11-17. doi: 10.1016/j.vetmic.2018.06.011.
doi: S0378-1135(17)31466-9 pmid: 30080665 |
| [31] |
KIMBLE J B, SORRELL E, SHAO H X, MARTIN P L, PEREZ D R. Compatibility of H9N2 avian influenza surface genes and 2009 pandemic H1N1 internal genes for transmission in the ferret model. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(29): 12084-12088. doi: 10.1073/pnas.1108058108.
doi: 10.1073/pnas.1108058108 pmid: 21730147 |
| [32] |
HU Z B, PENG F H, XIONG Z H, ZHANG W P, LI T T, SHI Y J, XIE J, JIN X, HUANG J J, XIAO H D, BI D R, SONG N H, LI Z L. Genetic and molecular characterization of H9N2 avian influenza viruses isolated from live poultry markets in Hubei Province, central China, 2013-2017. Virologica Sinica, 2021, 36(2): 291-299. doi: 10.1007/s12250-020-00260-z.
doi: 10. 1007/s12250-020-00260-z |
| [33] |
庄丽, 王恒, 冯蓓, 蒋琳, 蒋维佳. 贵州省首次报道人感染H9N2禽流感病例的病原学诊断及意义. 贵州医药, 2019, 43(4): 617-618. doi: 10.3969/j.issn.1000-744X.2019.04.044.
doi: 10.3969/j.issn.1000-744X.2019.04.044 |
|
ZHUANG L, WANG H, FENG B, JIANG L, JIANG W J. The etiological diagnosis and significance of a human case of H9N2 avian influenza reported in Guizhou Province for the first time. Guizhou Medical Journal, 2019, 43(4): 617-618. doi: 10.3969/j.issn.1000-744X.2019.04.044. (in Chinese)
doi: 10.3969/j.issn.1000-744X.2019.04.044 |
|
| [34] |
朱汝南, 孙宇, 王芳, 赵林清, 邓洁, 田润, 钱渊. 北京儿童中发现人感染禽流感病毒H9N2病例一例. 中华儿科杂志, 2017, 55(1): 69. doi: 10.3760/cma.j.issn.0578-1310.2017.01.016.
doi: 10.3760/cma.j.issn.0578-1310.2017.01.016 |
|
ZHU R N, SUN Y, WANG F, ZHAO L Q, DENG J, TIAN R, QIAN Y. A case of human infection with avian influenza virus H9N2 was found in a Beijing child. Chinese Journal of Pediatrics, 2017, 55(1): 69. doi: 10.3760/cma.j.issn.0578-1310.2017.01.016. (in Chinese)
doi: 10.3760/cma.j.issn.0578-1310.2017.01.016 |
|
| [35] |
张斯钰, 黄一伟, 胡世雄, 张恒娇, 孙倩莱, 邓志红, 曾舸, 张红, 湛志飞, 高立冬. 湖南省2005—2017年人感染禽流感流行病学特征分析. 中华疾病控制杂志, 2018, 22(10): 1037-1040. doi: 10.16462/j.cnki.zhjbkz.2018.10.014.
doi: 10. 16462/j.cnki.zhjbkz.2018.10.014 |
|
ZHANG S Y, HUANG Y W, HU S X, ZHANG H J, SUN Q L, DENG Z H, ZENG G, ZHANG H, ZHAN Z F, GAO L D. Epidemiologic characteristics of human avian influenza in Hunan Province from 2005 to 2017. Chinese Journal of Disease Control & Prevention, 2018, 22(10): 1037-1040. doi: 10.16462/j.cnki.zhjbkz.2018.10.014. (in Chinese)
doi: 10. 16462/j.cnki.zhjbkz.2018.10.014 |
|
| [36] |
杨磊, 杜训波, 张晓春, 岳勇, 翁贵武, 昝宇, 韩德琳. 成都市首例人感染H9N2禽流感病例调查与分析. 中国人兽共患病学报, 2017, 33(3): 245-249. doi: 10.3969/j.issn.1002-2694.2017.03.010.
doi: 10.3969/j.issn.1002-2694.2017.03.010 |
|
YANG L, DU X B, ZHANG X C, YUE Y, WENG G W, ZAN Y, HAN D L. Investigation and analysis of the first cases of human infection with avian influenza A(H9N2) virus in Chengdu, China. Chinese Journal of Zoonoses, 2017, 33(3): 245-249. doi: 10.3969/j.issn.1002-2694.2017.03.010. (in Chinese)
doi: 10.3969/j.issn.1002-2694.2017.03.010 |
|
| [37] |
罗春蕊, 赵晓南, 宁德明, 李多, 徐闻. 云南省首例人感染H9N2禽流感病例发现与应对. 中国人兽共患病学报, 2017, 33(3): 241-244. doi: 10.3969/j.issn.1002-2694.2017.03.009.
doi: 10.3969/j.issn.1002-2694.2017.03.009 |
|
LUO C R, ZHAO X N, NING D M, LI D, XU W. Discovery and response of the first case of human infection with avian influenza A(H9N2) virus in Yunnan Province, China. Chinese Journal of Zoonoses, 2017, 33(3): 241-244. doi: 10.3969/j.issn.1002-2694.2017.03.009. (in Chinese)
doi: 10.3969/j.issn.1002-2694.2017.03.009 |
|
| [38] |
刘峰, 李刚, 刘凤仁, 俞国龙. 广东省深圳市首例孕妇感染H9N2禽流感病例流行病学调查. 疾病监测, 2019, 34(7): 621-625. doi: 10.3784/j.issn.1003-9961.2019.07.010.
doi: 10.3784/j.issn.1003-9961.2019.07.010 |
|
LIU F, LI G, LIU F R, YU G L. Epidemiological survey of the first case of pregnant women infection with avian influenza A (H9N2) virus in Shenzhen. Disease Surveillance, 2019, 34(7): 621-625. doi: 10.3784/j.issn.1003-9961.2019.07.010. (in Chinese)
doi: 10.3784/j.issn.1003-9961.2019.07.010 |
|
| [39] |
黄政, 欧新华, 袁洁, 刘晓蕾. 长沙市首例人感染H9N2禽流感病毒的HA基因序列特征分析. 中国人兽共患病学报, 2016, 32(11): 997-1000. doi: 10.3969/j.issn.1002-2694.2016.011.010.
doi: 10.3969/j.issn.1002-2694.2016.011.010 |
|
HUANG Z, OU X H, YUAN J, LIU X L. Isolation and characteristic analysis of an avian influenza virus (H9N2) from a patient in Changsha, China. Chinese Journal of Zoonoses, 2016, 32(11): 997-1000. doi: 10.3969/j.issn.1002-2694.2016.011.010. (in Chinese)
doi: 10.3969/j.issn.1002-2694.2016.011.010 |
|
| [40] | 何军, 刘丽萍, 侯赛, 龚磊, 吴家兵, 胡万富, 王建军. 安徽省2株人感染H9N2流感病毒基因特征. 中华流行病学杂志, 2016, 37(5): 708-713. |
| HE J, LIU L P, HOU S, GONG L, WU J B, HU W F, WANG J J. Genetic characteristics of two human H9N2 influenza virus strains in Anhui Province. Chinese Journal of Epidemiology, 2016, 37(5): 708-713. (in Chinese) | |
| [41] | 郭元吉, 李建国, 程小雯, 王敏, 邹毅, 李钏华, 蔡访潺, 廖华乐, 张烨, 郭俊峰, 黄瑞敏, 贝东. 禽H9N2亚型流感病毒能感染人的发现. 中华实验和临床病毒学杂志, 1999, 13(2): 105. |
| GUO Y J, LI J G, CHENG X W, WANG M, ZOU Y, LI C H, CAI F C, LIAO H L, ZHANG Y, GUO J F, HUANG R M, BEI D. Discovery of men infected by avian influenza A(H9N2) virus. Chinese Journal of Experimental and Clinical Virology, 1999, 13(2): 105. (in Chinese) | |
| [42] |
BUTT K M, SMITH G J D, CHEN H L, ZHANG L J, LEUNG Y H C, XU K M, LIM W, WEBSTER R G, YUEN K Y, PEIRIS J S M, GUAN Y. Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. Journal of Clinical Microbiology, 2005, 43(11): 5760-5767. doi: 10.1128/JCM.43.11.5760-5767.2005.
doi: 10.1128/JCM.43.11.5760-5767.2005 pmid: 16272514 |
| [43] |
SONG W J, QIN K. Human-infecting influenza A (H9N2) virus: a forgotten potential pandemic strain? Zoonoses and Public Health, 2020, 67(3): 203-212. doi: 10.1111/zph.12685.
doi: 10.1111/zph.12685 pmid: 31930694 |
| [44] |
DONG X, XIONG J S, HUANG C L, XIANG J, WU W J, CHEN N S, WEN D N, TU C, QIAO X L, KANG L, YAO Z Z, ZHANG D Y, CHEN Q J. RETRACTED ARTICLE: human H9N2 avian influenza infection: epidemiological and clinical characterization of 16 cases in China. Virologica Sinica, 2021, 36(3): 564. doi: 10.1007/s12250-020-00248-9.
doi: 10.1007/s12250-020- 00248-9 |
| [45] |
尹馨, 马树杰, 李梅, 邓国华, 侯玉杰, 崔鹏飞, 施建忠, 陈化兰. PB2蛋白E627V突变可增强H7N9病毒对小鼠的致病力. 中国农业科学, 2018, 51(17): 3379-3388. doi: 10.3864/j.issn.0578-1752.2018.17.012.
doi: 10.3864/j.issn.0578-1752. 2018.17.012 |
|
YIN X, MA S J, LI M, DENG G H, HOU Y J, CUI P F, SHI J Z, CHEN H L. Amino acid substitutions of E627V in polymerase basic protein 2 gene increases the pathogenicity of the H7N9 influenza virus in mice. Scientia Agricultura Sinica, 2018, 51(17): 3379-3388. doi: 10.3864/j.issn.0578-1752.2018.17.012. (in Chinese)
doi: 10.3864/j.issn.0578-1752. 2018.17.012 |
|
| [46] |
HUANG Y Y, HU B X, WEN X T, CAO S J, GAVRILOV B K, DU Q J, KHAN M I, ZHANG X M. Diversified reassortant H9N2 avian influenza viruses in chicken flocks in northern and Eastern China. Virus Research, 2010, 151(1): 26-32. doi: 10.1016/j.virusres.2010.03.010.
doi: 10.1016/j.virusres.2010.03.010 pmid: 20347894 |
| [47] | 乐高钟, 吴小秧, 易爱兰, 罗勤, 冯秀, 游胜. 江西省首例儿童禽流感H9N2亚型病例的调查分析. 现代预防医学, 2017, 44(5): 785-787, 792. |
| YUE G Z, WU X Y, YI A L, LUO Q, FENG X, YOU S. The first case of children & #39;s H9N2 subtype avian influenza in Jiangxi Province. Modern Preventive Medicine, 2017, 44(5): 785-787, 792. (in Chinese) | |
| [48] |
PUSCH E A, SUAREZ D L. The multifaceted zoonotic risk of H9N2 avian influenza. Veterinary Sciences, 2018, 5(4): 82. doi: 10.3390/vetsci5040082.
doi: 10.3390/ vetsci5040082 |
| [49] |
ZHANG P H, TANG Y H, LIU X W, LIU W B, ZHANG X R, LIU H Q, PENG D X, GAO S, WU Y T, ZHANG L Y, LU S, LIU X F. A novel genotype H9N2 influenza virus possessing human H5N1 internal genomes has been circulating in poultry in Eastern China since 1998. Journal of Virology, 2009, 83(17): 8428-8438. doi: 10.1128/JVI.00659-09.
doi: 10.1128/JVI.00659-09 pmid: 19553328 |
| [50] |
LIU H Q, LIU X F, CHENG J, PENG D X, JIA L J, HUANG Y. Phylogenetic analysis of the hemagglutinin genes of twenty-six avian influenza viruses of subtype H9N2 isolated from chickens in China during 1996-2001. Avian Diseases, 2003, 47(1): 116-127. doi: 10.1637/0005-2086(2003)047[0116:PAOTHG]2.0.CO;2.
doi: 10. 1637/0005-2086(2003)047[0116: PAOTHG]2.0.CO pmid: 12713166 |
| [51] |
ZHU R, XU D W, YANG X Q, ZHANG J J, WANG S F, SHI H Y, LIU X F. Genetic and biological characterization of H9N2 avian influenza viruses isolated in China from 2011 to 2014. PLoS ONE, 2018, 13(7): e0199260. doi: 10.1371/journal.pone.0199260.
doi: 10.1371/journal.pone.0199260 |
| [52] |
LI X Y, SHI J Z, GUO J, DENG G H, ZHANG Q Y, WANG J L, HE X J, WANG K C, CHEN J M, LI Y Y, FAN J, KONG H, GU C Y, GUAN Y T, SUZUKI Y, KAWAOKA Y, LIU L L, JIANG Y P, TIAN G B, LI Y B, BU Z G, CHEN H L. Genetics, receptor binding property, and transmissibility in mammals of naturally isolated H9N2 Avian Influenza viruses. PLoS Pathogens, 2014, 10(11): e1004508. doi: 10.1371/journal.ppat.1004508.
doi: 10. 1371/journal.ppat.1004508 |
| [53] |
LIU X K, YANG C, SUN X M, LIN X, ZHAO L Z, CHEN H C, JIN M L. Evidence for a novel mechanism of influenza A virus host adaptation modulated by PB2-627. The FEBS Journal, 2019, 286(17): 3389-3400. doi: 10.1111/febs.14867.
doi: 10.1111/febs.14867 |
| [54] |
张静, 陈立根, 张宇, 孙乾晋, 李如意, 白牧原, 蔺文成, 谢青梅. 1997—2015年我国不同地区H9N2亚型禽流感病毒流行情况研究. 中国家禽, 2016, 38(20): 20-27. doi: 10.16372/j.issn.1004-6364.2016.20.005.
doi: 10.16372/j.issn.1004-6364.2016. 20.005 |
|
ZHANG J, CHEN L G, ZHANG Y, SUN Q J, LI R Y, BAI M Y, LIN W C, XIE Q M. Prevalence of H9N2 subtype avian influenza virus in different regions of China during 1997 to 2015. China Poultry, 2016, 38(20): 20-27. doi: 10.16372/j.issn.1004-6364.2016.20.005. (in Chinese)
doi: 10.16372/j.issn.1004-6364.2016. 20.005 |
|
| [55] |
JIAO P R, TIAN G B, LI Y B, DENG G H, JIANG Y P, LIU C, LIU W L, BU Z G, KAWAOKA Y, CHEN H L. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. Journal of Virology, 2008, 82(3): 1146-1154. doi: 10.1128/jvi.01698-07.
doi: 10.1128/jvi.01698-07 |
| [56] |
FAN S F, DENG G H, SONG J S, TIAN G B, SUO Y B, JIANG Y P, GUAN Y T, BU Z G, KAWAOKA Y, CHEN H L. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology, 2009, 384(1): 28-32. doi: 10.1016/j.virol.2008.11.044.
doi: 10.1016/j.virol.2008.11.044 pmid: 19117585 |
| [57] | 王云鹤, 包红梅, 孙佳善, 李雁冰, 徐晓龙, 王子龙, 施建忠, 曾显营, 王秀荣, 陈化兰. H7N9亚型禽流感病毒RT-PCR检测方法建立. 中国农业科学, 2015, 48(15): 3050-3055. |
| WANG Y H, BAO H M, SUN J S, LI Y B, XU X L, WANG Z L, SHI J Z, ZENG X Y, WANG X R, CHEN H L. Development of RT-PCR technique for detection of H7N9 subtype avian influenza virus. Scientia Agricultura Sinica, 2015, 48(15): 3050-3055. (in Chinese) | |
| [58] |
王萍萍, 郭晶, 李玉保, 刘成, 李旭勇. H7N9亚型禽流感病毒研究进展. 中国人兽共患病学报, 2021, 37(2): 159-164, 170. doi: 10.3969/j.issn.1002-2694.2021.00.010.
doi: 10. 3969/j.issn.1002-2694.2021.00.010 |
|
WANG P P, GUO J, LI Y B, LIU C, LI X Y. Research progress in H7N9 subtype avian influenza viruses. Chinese Journal of Zoonoses, 2021, 37(2): 159-164, 170. doi: 10.3969/j.issn.1002-2694.2021.00.010. (in Chinese)
doi: 10. 3969/j.issn.1002-2694.2021.00.010 |
|
| [59] |
辛丽, 白天, 周剑芳, 唐静, 陈永坤, 陈涛, 史景红, 李晓丹, 李燕. 中国职业暴露人群感染H9N2禽流感病毒血清学调查. 疾病监测, 2015, 30(5): 368-371. doi: 10.3784/j.issn.1003-9961.2015.05.007.
doi: 10.3784/j.issn.1003-9961.2015.05.007 |
|
XIN L, BAI T, ZHOU J F, TANG J, CHEN Y K, CHEN T, SHI J H, LI X D, LI Y. Serological study of human infection with avian influenza A (H9N2) virus in population with occupational exposure in China. Disease Surveillance, 2015, 30(5): 368-371. doi: 10.3784/j.issn.1003-9961.2015.05.007. (in Chinese)
doi: 10.3784/j.issn.1003-9961.2015.05.007 |
| [1] | 缪葭皓, 崔鹏飞, 颜成, 王丛丛, 王燕, 陈源, 陈鹏, 施建忠, 邓国华, 陈化兰. 鸭源H8N4亚型禽流感病毒的遗传演化及其部分生物学特性分析[J]. 中国农业科学, 2026, 59(7): 1576-1586. |
| [2] | 陈源, 崔鹏飞, 施建忠, 张元成, 于晴晴, 颜成, 张亚萍, 王丛丛, 张洁, 王燕, 邓国华, 陈化兰. 2019-2022年中国H6N1亚型禽流感病毒的生物学特性分析[J]. 中国农业科学, 2024, 57(9): 1820-1832. |
| [3] | 李博天, 李春琪, 刘国平, 谢军, 曾攀, 赵润泽, 李桐, 裴洁, 郭利伟, 伍锐, 谭磊. 2020-2022年华中地区部分猪场猪圆环病毒3型的分子流行特征与遗传变异分析[J]. 中国农业科学, 2024, 57(3): 613-626. |
| [4] | 罗素贤, 周红, 蔺辉星, 范红结. 我国中东部主要养猪地区死亡育肥猪呼吸道病原菌的流行病学调查及猪多杀性巴氏杆菌的特性鉴定[J]. 中国农业科学, 2024, 57(11): 2254-2264. |
| [5] | 麻琦, 和新文, 王燕, 刘艳晶, 潘舒心, 侯玉杰, 施建忠, 邓国华, 包红梅, 刘景利, 郭兴福, 毛胜刚, 胡井雷, 路通, 杨帆, 田国彬, 曾显营, 陈化兰. 商品疫苗对我国h9.4.2.5分支H9N2亚型禽流感分离株的免疫保护[J]. 中国农业科学, 2023, 56(15): 3040-3050. |
| [6] | 邵震, 刁有祥. 规模化鹅场主要病毒核酸检测结果与分析[J]. 中国农业科学, 2023, 56(10): 2021-2034. |
| [7] | 杨程,龚桂芝,彭祝春,常珍珍,易璇,洪棋斌. 基于cpInDel标记和cpSSR标记的柑橘属及近缘属植物亲缘关系[J]. 中国农业科学, 2022, 55(16): 3210-3223. |
| [8] | 周群,陈小飞,阚蕊慈,李玉,曹慧,彭艳伶,张斌. 2017-2019年四川地区猪A群轮状病毒的分子流行病学调查[J]. 中国农业科学, 2021, 54(5): 1063-1072. |
| [9] | 孙莹,张兵,李岭,黄小洁,侯力丹,刘丹,李启红,李俊平,王乐元,李慧姣,杨承槐. 表达H9亚型禽流感病毒HA基因重组鸭肠炎病毒的构建[J]. 中国农业科学, 2019, 52(23): 4398-4405. |
| [10] | 罗玉子,孙元,王涛,仇华吉. 非洲猪瘟——我国养猪业的重大威胁[J]. 中国农业科学, 2018, 51(21): 4177-4187. |
| [11] | 李莉,杜鑫,张丽娜,杨柳,高晓庆,唐东雪,赵海源,蒋晓梅,张天舒,李金祥. 重组禽流感病毒H7N9 H7-Re1株培养条件的优化[J]. 中国农业科学, 2018, 51(17): 3415-3426. |
| [12] | 陈宏,杨柳,宋海岩,石莹,孟令伟,付春杰,张丹,赵海源,李金祥,蒋晓梅,张天舒. 悬浮MDCK细胞的驯化与H5亚型禽流感病毒的培养[J]. 中国农业科学, 2018, 51(17): 3405-3414. |
| [13] | 杨劲松,吴涛,李金祥. 重组禽流感病毒H5亚型灭活疫苗的质量分析及发展方向[J]. 中国农业科学, 2018, 51(17): 3397-3404. |
| [14] | 王云鹤,包红梅,孙佳善,李雁冰,徐晓龙,王子龙,施建忠,曾显营,王秀荣,陈化兰. H7N9亚型禽流感病毒RT-PCR检测方法建立[J]. 中国农业科学, 2015, 48(15): 3050-3055. |
| [15] | 赵晴晴,李群辉,朱杰,钟蕾,刘晶晶,顾敏,王晓泉,刘文博,刘秀梵. 一株鸭源H4N8亚型禽流感病毒的全基因测序及遗传进化分析[J]. 中国农业科学, 2015, 48(15): 3040-3049. |
|
||