






中国农业科学 ›› 2025, Vol. 58 ›› Issue (1): 75-90.doi: 10.3864/j.issn.0578-1752.2025.01.006
曹言勇1( ), 程泽强1(), 马娟1, 杨文博1, 朱卫红1, 孙新艳1, 李慧敏1, 夏来坤1,*(), 段灿星2,*()
), 程泽强1(), 马娟1, 杨文博1, 朱卫红1, 孙新艳1, 李慧敏1, 夏来坤1,*(), 段灿星2,*()
收稿日期:2024-07-28
接受日期:2024-09-24
出版日期:2025-01-01
发布日期:2025-01-07
通信作者:
联系方式:
曹言勇,E-mail:yanyongcao@126.com。程泽强,E-mail:zeqiangcheng@163.com。曹言勇和程泽强为同等贡献作者。
基金资助:
CAO YanYong1(), CHENG ZeQiang1(), MA Juan1, YANG WenBo1, ZHU WeiHong1, SUN XinYan1, LI HuiMin1, XIA LaiKun1,*(), DUAN CanXing2,*()
Received:2024-07-28
Accepted:2024-09-24
Published:2025-01-01
Online:2025-01-07
摘要:
【目的】玉米茎腐病严重威胁玉米的产量与品质,其致病菌复杂,层出镰孢(Fusarium proliferatum)近年来逐渐成为主要病原之一。本研究旨在通过多组学联合分析,深入探究玉米对层出镰孢茎腐病的响应机制,明确差异基因和差异代谢物富集的信号通路在玉米抗病过程中的关键作用,为玉米抗茎腐病育种和病害防控提供理论依据。【方法】选择对层出镰孢具有不同抗性的玉米自交系ZC17(抗病)和CH72(感病)作为研究材料。在玉米9叶期,对其进行接种处理,接种组注射层出镰孢菌液,模拟接种组注射等量的PDB,随后用凡士林封闭伤口。接种后7 d,采集接种区域上下茎段中间位置的组织样本,分别用于转录组测序和非靶向代谢组学检测。同时对接种后的植株进行茎腐病症状评估,计算茎腐病平均评分(SRSA)和病情指数(DSI)。利用多种生物信息学工具对转录组测序数据和代谢组学数据进行分析,并通过实时荧光定量PCR(RT-qPCR)对差异基因进行验证。【结果】表型和生理数据显示,接种层出镰孢后,CH72发病程度显著高于ZC17,其SRSA增加2.48倍,DSI增加35.36%。转录组和代谢组的PCA结果显示,各组内样本的重现性很高,ZC17和CH72相互分离,FP组和MK组相互分离。转录组分析表明,接种后CH72的差异表达基因数量多于ZC17,但二者近50%的差异基因表达趋势相同。功能注释和富集分析发现,差异基因和差异代谢物主要富集于植物次生代谢产物生物合成、苯丙氨酸代谢、植物激素生物合成及植物-病原体相互作用等途径。转录组和代谢组联合分析证实,苯丙氨酸代谢以及苯丙氨酸、酪氨酸和色氨酸生物合成在玉米抗层出镰孢过程中起关键作用。此外,多个转录因子家族(如MYB、bHLH、NAC和WRKY等)在接种层出镰孢后被显著激活,表明这些转录因子在玉米抗病分子调控网络中可能发挥重要作用。qPCR与转录组测序结果在4个组中的表达趋势一致,Spearman相关分析也显示转录组测序数据和qPCR结果之间高度一致(r=0.75,P=7.5e-05)。【结论】苯丙氨酸代谢相关途径在玉米响应层出镰孢茎腐病中至关重要;C4H、PAL、ADT、GOT等关键酶以及显著上调的代谢物如2-香豆酸、3-羟基肉桂酸、吲哚、苯丙氨酸、色氨酸和酪氨酸在植物抗病中起重要作用。本研究挖掘出的潜在抗病相关转录因子、基因和代谢物可为深入解析玉米对层出镰孢茎腐病的分子响应机制提供重要依据。
曹言勇, 程泽强, 马娟, 杨文博, 朱卫红, 孙新艳, 李慧敏, 夏来坤, 段灿星. 整合转录组和代谢组学分析揭示玉米对层出镰孢茎腐病的响应机制[J]. 中国农业科学, 2025, 58(1): 75-90.
CAO YanYong, CHENG ZeQiang, MA Juan, YANG WenBo, ZHU WeiHong, SUN XinYan, LI HuiMin, XIA LaiKun, DUAN CanXing. Integrating Transcriptomic and Metabolomic Analyses Reveals Maize Responses to Stalk Rot Caused by Fusarium proliferatum[J]. Scientia Agricultura Sinica, 2025, 58(1): 75-90.
表1
用于RT-qPCR验证的基因及引物序列"
| 基因 Gene | 引物Primer | 序列 Sequence (5′ to 3′) |
|---|---|---|
| Zm00001d028815 | 8815-1F | GACGCCTACAACTAAACC |
| 8815-1R | ACAACACAAGGACAGCAC | |
| Zm00001d028816 | 8816-1F | GATTTCTTCCTTGCCTTTT |
| 8816-1R | ACACACCCACACACATTTG | |
| Zm00001d028313 | 8313-2F | AAAGGAGGAAGGAGACCA |
| 8313-2R | CCCGCCATAGAAGATTGT | |
| Zm00001d014250 | 4250F | AAGTGTCGCTGCAGAGGTTC |
| 4250R | TCCTTTAAGGCCGTCGCTTG | |
| Zm00001d022139 | 2139F | CCGACAAGGCAAAGGATCTG |
| 2139R | GGCTCTCAGGCTCCTTCTTG | |
| Zm00001d043528 | 3528-1F | AGCTTGCCCCTTTCTCAAC |
| 3528-1R | ACCAGGAACACCGTCATTT | |
| Zm00001d017121 | 7121F | CCAATGCTAGCTGCACAACC |
| 7121R | AACAGCCTTAGCAGCTCCAG | |
| Zm00001d004366 | 4366F | ACTCATTCCGCGACATCGAA |
| 4366R | GCGGTCATCGTCAAGGTACA | |
| Zm00001d043144 | 3144F | ACGACGAGTTCGTCAAGGTC |
| 3144R | TGCACCAGCATGTACTGGAC | |
| Zm00001d021168 | 1168F | CCAGGTTTCCACCGTGCTTC |
| 1168R | AGGACACGTACAGGACGGAC | |
| Zm00001d009028 | 9028F | TTGAACTGCGCCGCCTTG |
| 9028R | AGGAAACCCACAGACTTGGC | |
| GAPDH | GAPDH1F | CCATCACTGCCACACAGAAAAC |
| GAPDH1R | AGGAACACGGAAGGACATACCAG |
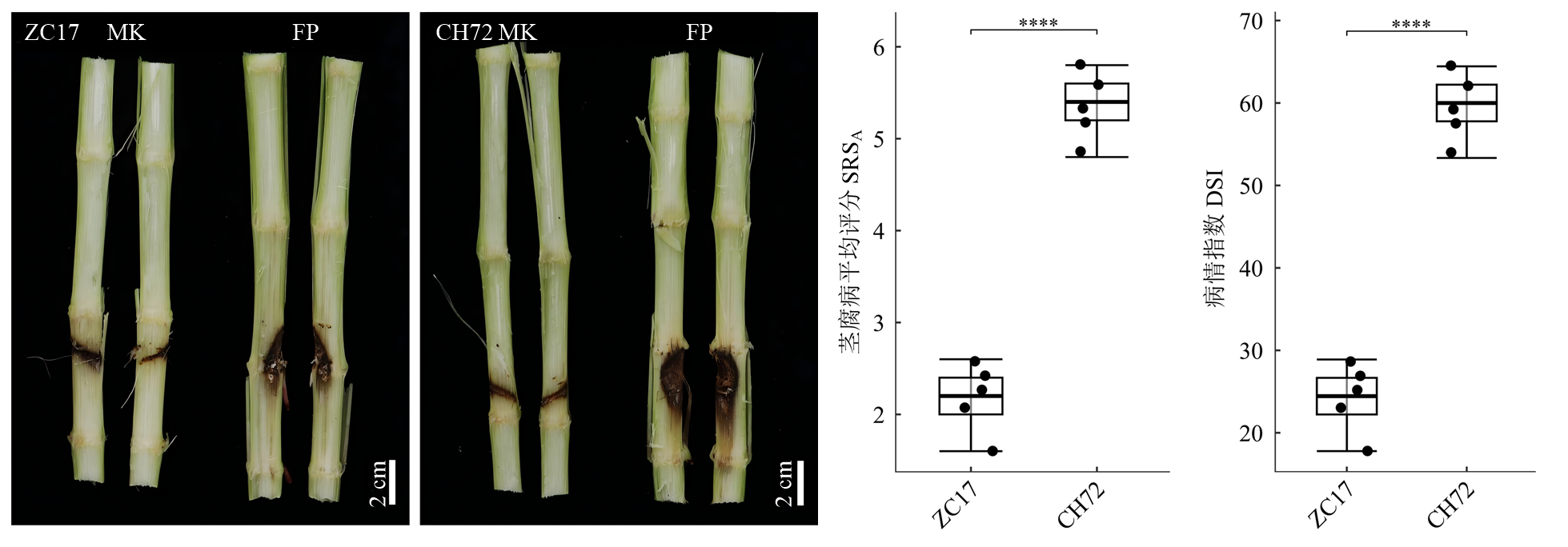
图1
不同抗性玉米近交系接种后的表型和病害症状"
表2
转录组测序数据总览"
| 样品 Sample | 原始序列数 Raw reads | 原始碱基数 Raw bases | 有效序列数 Valid reads | 有效碱基数 Valid bases | 有效比率 Valid ratio (%) | Q20 (%) | Q30 (%) | 基因组比对序列数Mapped reads |
|---|---|---|---|---|---|---|---|---|
| ZC17_MK1 | 48487174 | 7.27G | 46461886 | 6.97G | 95.82 | 99.94 | 97.59 | 41846591 (90.07%) |
| ZC17_MK2 | 38471678 | 5.77G | 37250738 | 5.59G | 96.83 | 99.95 | 98.99 | 33782858 (90.69%) |
| ZC17_MK3 | 42311182 | 6.35G | 40990978 | 6.15G | 96.88 | 99.95 | 98.79 | 37004587 (90.27%) |
| ZC17_FP1 | 44341730 | 6.65G | 43314250 | 6.50G | 97.68 | 99.95 | 98.69 | 39449341 (91.08%) |
| ZC17_FP2 | 44878826 | 6.73G | 43756214 | 6.56G | 97.50 | 99.95 | 98.70 | 39796076 (90.95%) |
| ZC17_FP3 | 44490646 | 6.67G | 43362776 | 6.50G | 97.46 | 99.95 | 98.99 | 39559652 (91.23%) |
| CH72_MK1 | 48737466 | 7.31G | 47558210 | 7.13G | 97.58 | 99.95 | 98.37 | 42325490 (89.00%) |
| CH72_MK2 | 39150064 | 5.87G | 38093258 | 5.71G | 97.30 | 99.96 | 99.02 | 34198799 (89.78%) |
| CH72_MK3 | 45219372 | 6.78G | 44260068 | 6.64G | 97.88 | 99.95 | 98.68 | 39511099 (89.27%) |
| CH72_FP1 | 47658538 | 7.15G | 46484104 | 6.97G | 97.54 | 99.96 | 98.56 | 41481231 (89.24%) |
| CH72_FP2 | 43802340 | 6.57G | 42961890 | 6.44G | 98.08 | 99.96 | 99.17 | 38760342 (90.22%) |
| CH72_FP3 | 41930108 | 6.29G | 40712488 | 6.11G | 97.10 | 99.96 | 99.13 | 34565595 (84.90%) |
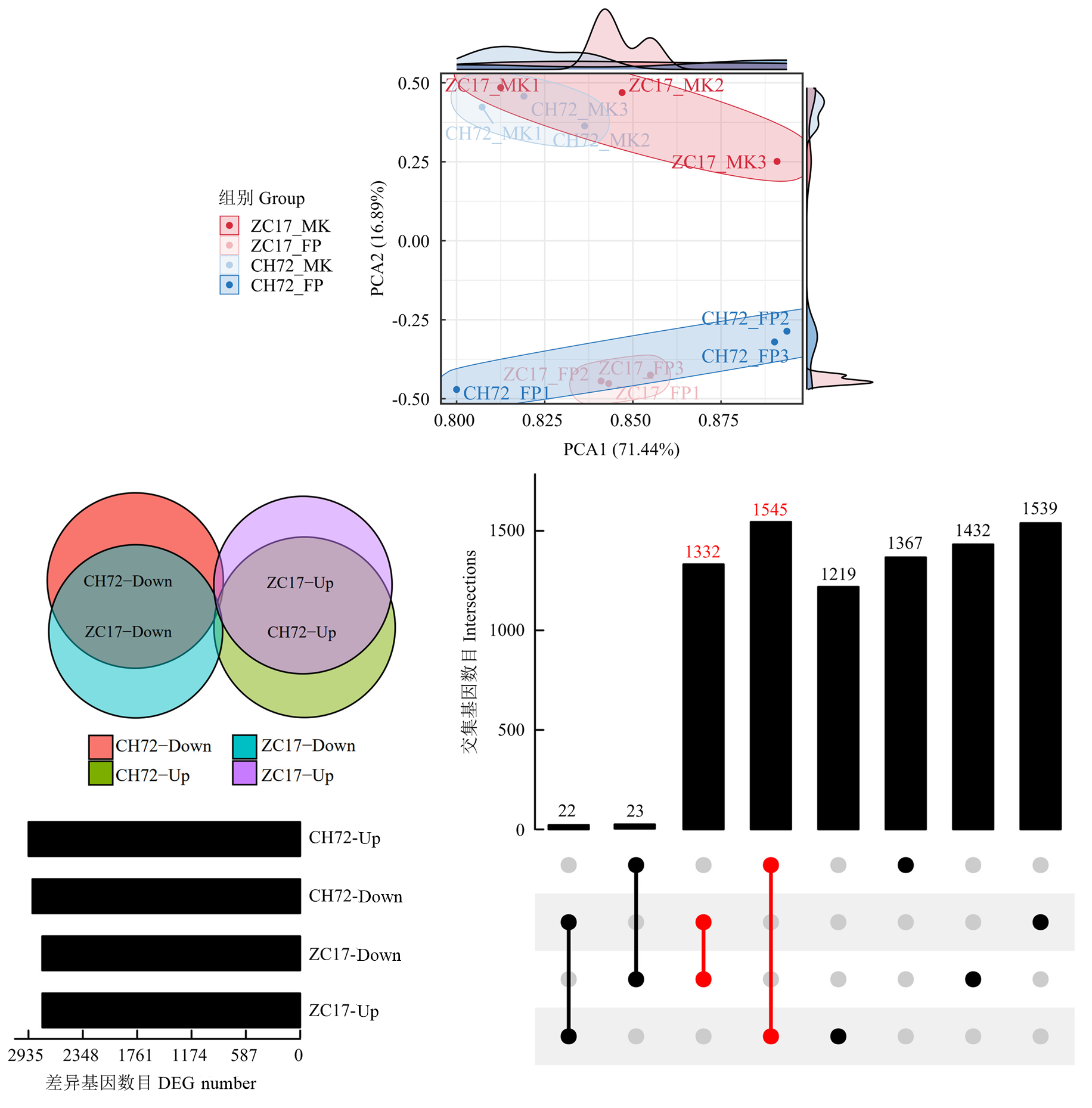
图2
不同抗性自交系样本的主成分分析及差异基因筛选"
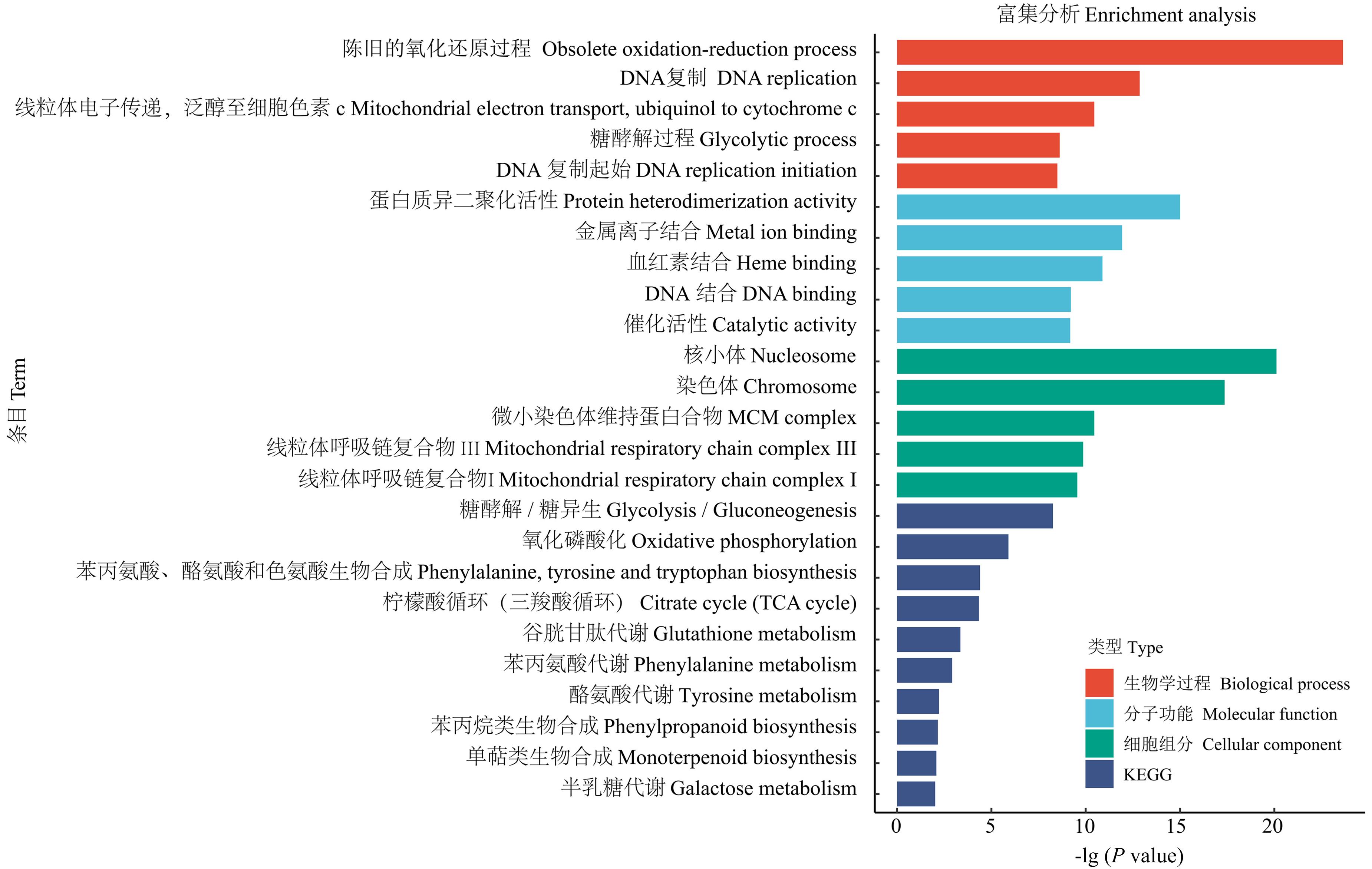
图3
玉米应答层出镰孢侵染的差异基因功能和通路富集分析"
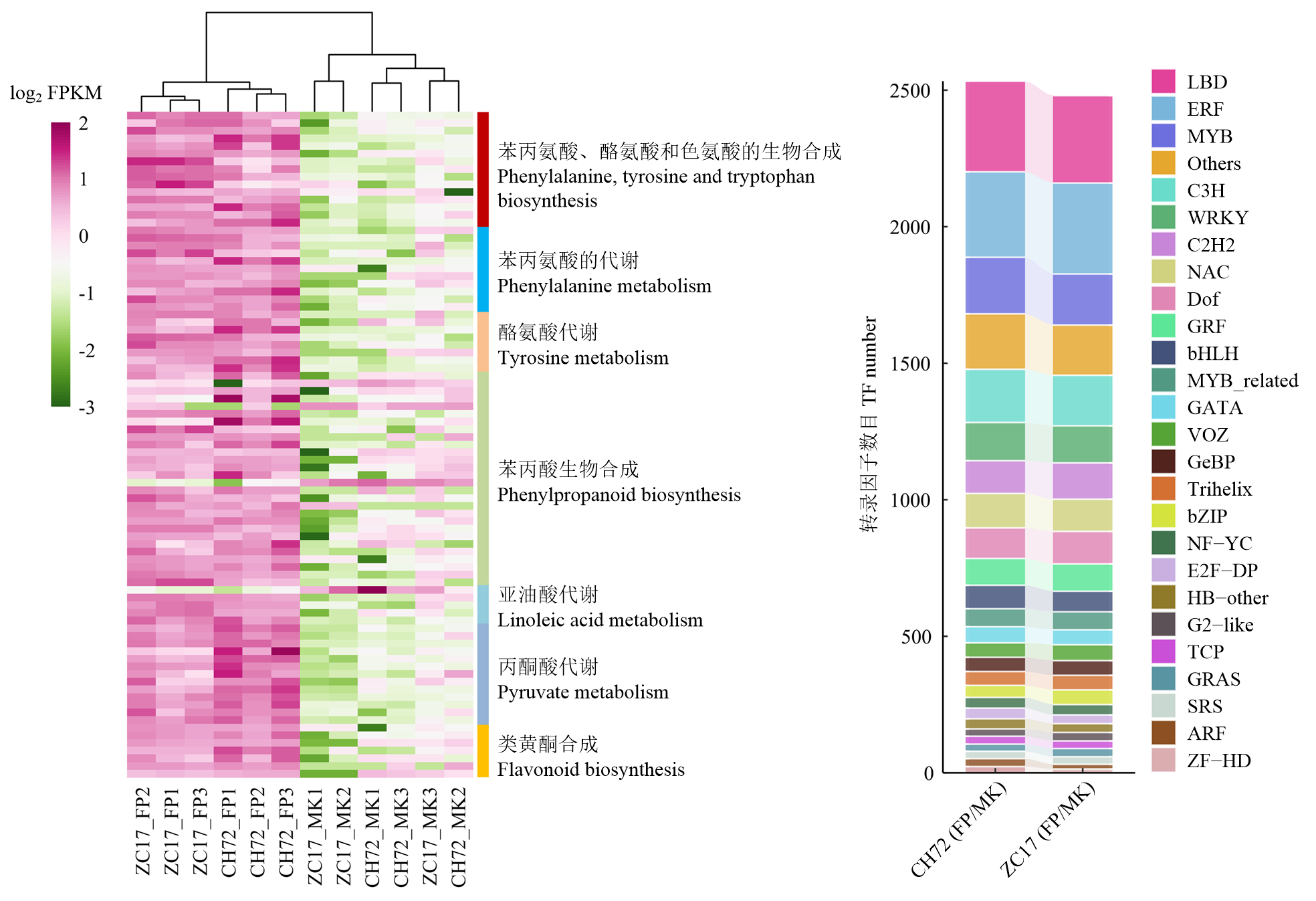
图4
植物主要抗病通路中差异基因的热图及转录因子鉴定"
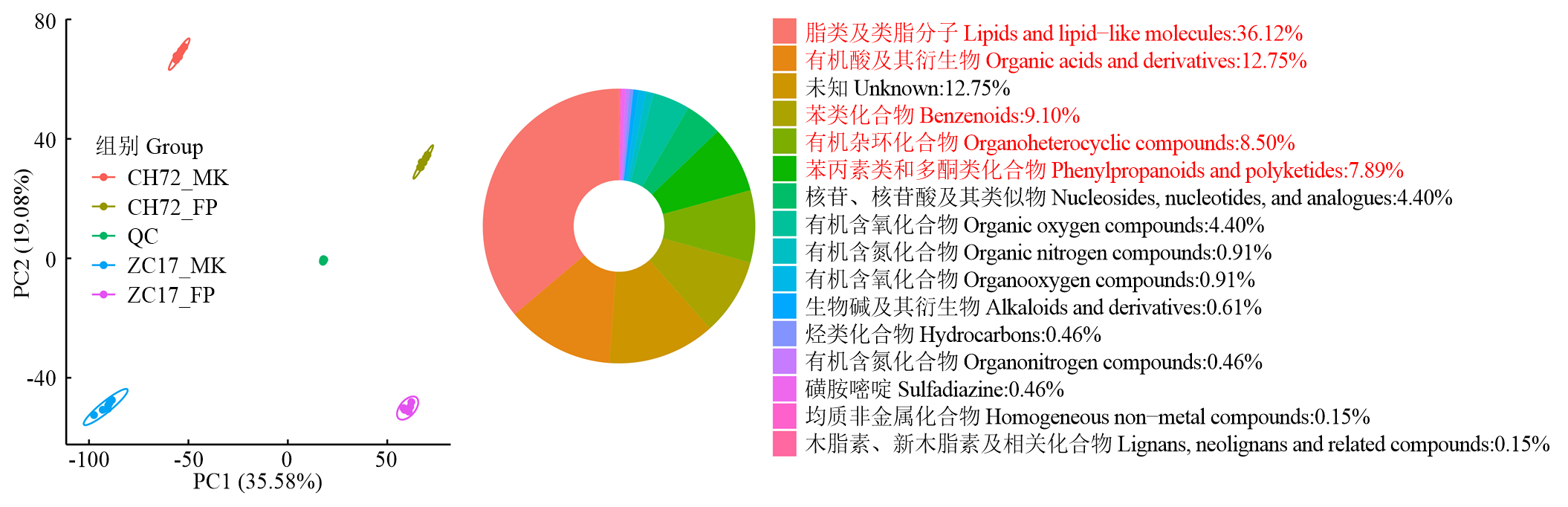
图5
代谢组样本主成分分析及代谢物鉴定"
表3
不同比较组中上调和下调差异最大的前10种代谢物"
| 比较组 Comparison | 代谢物 Metabolite | Log2FC |
|---|---|---|
| CH72 (FP/MK) | 蜀葵苷Scorzoside | 9.201 |
| N-(p-羟苯基)乙基对羟基肉桂酰胺N-(p-Hydroxyphenyl)ethyl p-hydroxycinnamide | 7.304 | |
| 伏马菌素B1 Fumonisin B1 | 6.862 | |
| 对香豆酸乙酯Ethyl p-coumarate | 6.373 | |
| L-苯丙氨酸L-Phenylalanine | 5.878 | |
| 肉桂酸Cinnamic acid | 5.759 | |
| 苯丙氨酸Phenylalanine | 5.666 | |
| N-(1-脱氧-1-果糖基)酪氨酸N-(1-Deoxy-1-fructosyl)tyrosine | 5.439 | |
| 吲哚-3-乙酰胺Indole-3-acetamide | 5.205 | |
| 溶血磷脂酰肌醇15:0 LysoPI 15:0 | 4.876 | |
| 1-硬脂酰-2-羟基-sn-甘油-3-磷酸胆碱1-Stearoyl-2-hydroxy-sn-glycero-3-phosphocholine | -4.631 | |
| 氧化型谷胱甘肽Glutathione, oxidized | -3.808 | |
| L-谷胱甘肽(氧化型)L-Glutathione (oxidized form) | -3.355 | |
| 2-(2-噻吩基)呋喃2-(2-Thienyl)furan | -3.311 | |
| 翠雀素-3-O-(6''-O-α-鼠李吡喃糖基-β-葡萄糖苷) Delphinidin-3-O-(6''-O-alpha-rhamnopyranosyl-beta-glucopyranoside) | -2.606 | |
| 溶血磷脂酰甘油16:0 LysoPG 16:0 | -2.547 | |
| 溶血磷脂酰胆碱18:1 LysoPC 18:1 | -2.251 | |
| 谷氨酰胺-亮氨酸Gln-Leu | -2.060 | |
| 缬氨酸-苯丙氨酸Val-Phe | -2.011 | |
| 酪氨酸-亮氨酸Tyr-Leu | -1.965 | |
| ZC17 (FP/MK) | 阿洛糖Allose | 6.719 |
| 乙酸Acetic acid | 6.535 | |
| 广木香内酯Costunolide | 5.894 | |
| D-1,5-脱水果糖D-1,5-Anhydrofructose | 5.306 | |
| 5-吡哆醇内酯5-Pyridoxolactone | 5.276 | |
| 伏马菌素B1 Fumonisin B1 | 5.182 | |
| 甘油1-十六酸酯Glycerol 1-hexadecanoate | 4.816 | |
| 溶血磷脂酰肌醇15:0 LysoPI 15:0 | 4.631 | |
| 棕矢车菊素Jaceidin | 4.175 | |
| 松油酸乙酯Pinolenic acid ethyl ester | 4.070 | |
| 溶血磷脂酰乙醇胺18:3 LysoPE 18:3 | -2.731 | |
| 2,4-二羟基苯甲酸2,4-Dihydroxybenzoic acid | -2.772 | |
| 缬氨酸-亮氨酸Val-Leu | -2.899 | |
| 前列腺素A1 Prostaglandin A1 | -3.159 | |
| 9-氢过氧基-10E,12Z,15Z-十八碳三烯酸9-Hydroperoxy-10E,12Z,15Z-octadecatrienoic acid | -3.160 | |
| 穆坪马兜铃酰胺Moupinamide | -3.722 | |
| (9S,10E,12S,13S)- 9,12,13-三羟基-10-十八碳烯酸(9S,10E,12S,13S)-9,12,13-Trihydroxy-10-octadecenoic acid | -3.923 | |
| 溶血磷脂酰胆碱18:3 LysoPC 18:3 | -3.984 | |
| (9ξ,10ξ,12ξ)- 9,10-二羟基-12-十八碳烯酸(9xi,10xi,12xi)-9,10-Dihydroxy-12-octadecenoic acid | -4.171 | |
| 去甲异波尔定Norisoboldine | -4.280 |
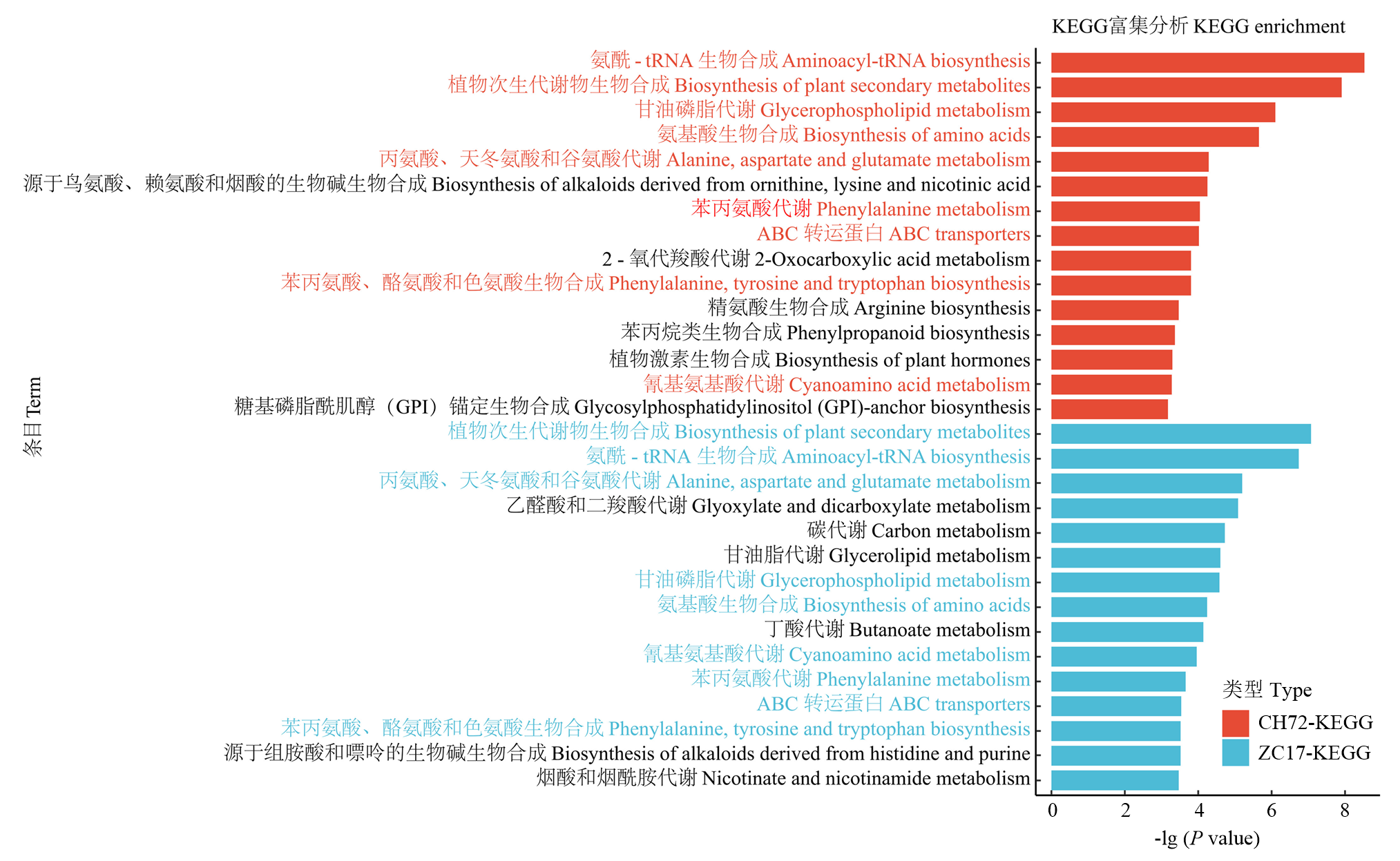
图6
差异代谢物KEGG通路富集分析"

图7
差异基因和差异代谢物的整合分析"
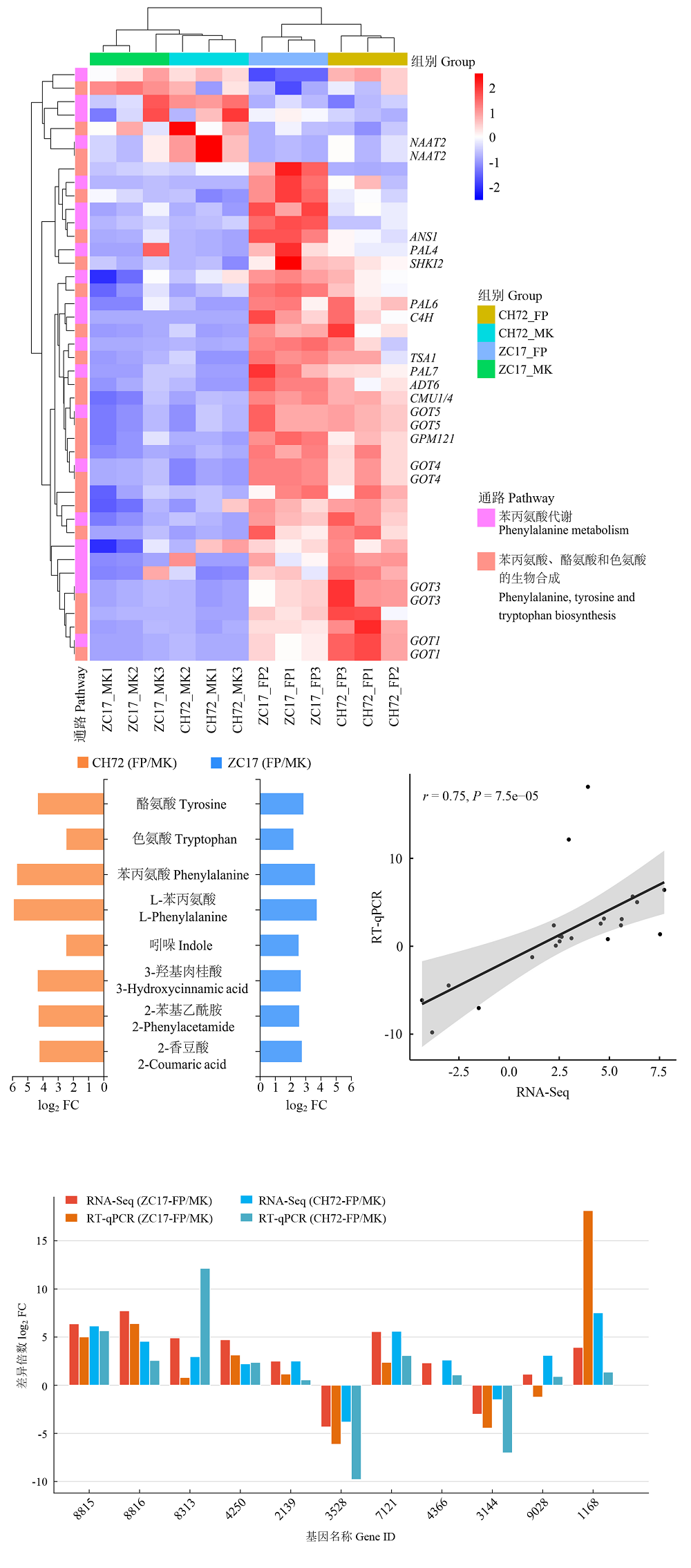
图8
苯丙氨酸、酪氨酸和色氨酸生物合成和苯丙氨酸代谢途径中关键基因和代谢物的分析及qPCR验证"
| [1] |
李少昆, 赵久然, 董树亭, 赵明, 李潮海, 崔彦宏, 刘永红, 高聚林, 薛吉全, 王立春, 王璞, 陆卫平, 王俊河, 杨祁峰, 王子明. 中国玉米栽培研究进展与展望. 中国农业科学, 2017, 50(11): 1941-1959. doi: 10.3864/j.issn.0578-1752.2017.11.001.
|
|
|
|
| [2] |
doi: 10.1007/s00122-010-1339-0 pmid: 20401458 |
| [3] |
段灿星, 曹言勇, 董怀玉, 夏玉生, 李红, 胡清玉, 杨知还, 王晓鸣. 玉米种质资源抗腐霉茎腐病和镰孢茎腐病精准鉴定. 中国农业科学, 2022, 55(2): 265-279. doi: 10.3864/j.issn.0578-1752.2022.02.003.
|
|
|
|
| [4] |
|
| [5] |
|
| [6] |
刘树森, 马红霞, 郭宁, 石洁, 张海剑, 孙华, 金戈. 黄淮海夏玉米主产区茎腐病主要病原菌及优势种分析. 中国农业科学, 2019, 52(2): 262-272. doi: 10.3864/j.issn.0578-1752.2019.02.006.
|
|
|
|
| [7] |
|
| [8] |
范志业, 崔小伟, 施艳, 陈琦, 刘迪, 侯艳红, 李世民, 闫海霞, 袁刘正, 孙虎. 河南省玉米茎基腐病主要病原菌鉴定及主栽玉米品种的抗性分析. 河南农业科学, 2014, 43(12): 87-90.
|
|
|
|
| [9] |
|
| [10] |
doi: 10.3390/ijms161226106 pmid: 26633370 |
| [11] |
doi: 10.1146/annurev-cellbio-092910-154055 pmid: 22559264 |
| [12] |
doi: 10.3389/fpls.2017.01774 pmid: 29075283 |
| [13] |
pmid: 15012548 |
| [14] |
doi: 10.1093/aob/mcg073 pmid: 12714366 |
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
doi: 10.1146/annurev-phyto-080516-035544 pmid: 28645231 |
| [21] |
doi: 10.1111/nph.12291 pmid: 24010138 |
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
刘春来. 中国玉米茎腐病研究进展. 中国农学通报, 2017, 33(30): 130-134.
doi: 10.11924/j.issn.1000-6850.casb16120102 |
|
doi: 10.11924/j.issn.1000-6850.casb16120102 |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
doi: 10.3390/molecules191016240 pmid: 25310150 |
| [33] |
doi: 10.1111/j.1469-8137.2009.03041.x pmid: 19807873 |
| [34] |
doi: 10.1146/annurev-arplant-042811-105439 pmid: 22554242 |
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
doi: S0092-8674(17)30649-9 pmid: 28666113 |
| [40] |
doi: 10.1016/j.tplants.2012.02.004 pmid: 22445067 |
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [1] | 岳丽昕, 王清华, 王振宝, 尼玛琼吉, 刘泽洲, 孔素萍, 张立峰, 高莉敏. 基于广靶代谢组学分析藏葱和细香葱营养品质及黄酮类代谢产物差异[J]. 中国农业科学, 2026, 59(5): 1070-1086. |
| [2] | 王忠妮, 雷月, 李佳丽, 宫彦龙, 朱速松. ABC转运蛋白OsARG1调控水稻抽穗期的功能[J]. 中国农业科学, 2026, 59(1): 1-16. |
| [3] | 王思琪, 邹利人, 白瑞雯, 闫可, 王思洋, 齐晓光, 申海林, 温景辉. 赤霉素调控‘蜜汁’葡萄穗轴硬化关键基因的挖掘[J]. 中国农业科学, 2026, 59(1): 179-189. |
| [4] | 张义茹, 韩雪, 姚鑫杰, 冯军, 魏爱丽, 李文超, 张彬, 韩渊怀, 李红英. 基于多组学解析谷子后熟米色变化的分子机制[J]. 中国农业科学, 2025, 58(9): 1702-1718. |
| [5] | 谭西北, 兰徐颖, 刘崇怀, 樊秀彩, 姜建福, 孙磊, 李鹏, 余书鑫, 张颖. 不同抗性葡萄响应白腐病侵染的次生代谢物变化[J]. 中国农业科学, 2025, 58(9): 1767-1778. |
| [6] | 吴郁, 曲翔汝, 杨丹, 伍芩, 陈国跃, 江千涛, 魏育明, 许强. 广泛非靶向代谢组学解析小麦抗条锈病反应中叶绿体代谢产物[J]. 中国农业科学, 2025, 58(7): 1333-1343. |
| [7] | 杨彩丽, 李永洲, 贺亮亮, 宋银花, 章鹏, 刘肇先, 李鹏慧, 刘三军. 葡萄TPS基因家族全基因组鉴定及VvTPS4在单萜形成中的功能验证[J]. 中国农业科学, 2025, 58(7): 1397-1417. |
| [8] | 邹晓威, 夏蕾, 朱晓敏, 孙辉, 周琦, 齐霁, 张亚封, 郑岩, 姜兆远. 基于转录组测序的玉米瘤黑粉菌UM01240过表达菌株诱导玉米抗病性分析[J]. 中国农业科学, 2025, 58(6): 1116-1130. |
| [9] | 孙萍, 朱文灿, 林贤锐, 吴嘉颀, 曹译文, 陈辰斐, 王轶, 朱建锡, 贾惠娟, 钱敏杰, 沈建生. 基于代谢组和转录组解析多雨寡照对桃果皮着色和类黄酮积累的影响[J]. 中国农业科学, 2025, 58(6): 1173-1194. |
| [10] | 谢露露, 李福, 张思远, 高建昌. 基于跨物种转录组解析影响不定根发生的保守基因[J]. 中国农业科学, 2025, 58(6): 1195-1209. |
| [11] | 高岩浩, 王婷婷, 白卫卫, 杜兴杰, 刘贤, 秦本源, 付彤, 孙宇, 高腾云, 张天留. 脂质组与转录组联合揭示南阳牛不同肌肉组织脂质特征的差异表达模式[J]. 中国农业科学, 2025, 58(6): 1239-1258. |
| [12] | 罗朝丹, 冯春梅, 李建强, 黎新荣, 韦勇, 杨李益, 刘筱瑾, 谭和, 任二芳, 罗小杰. 基于GC-MS非靶向代谢组学分析不同成熟度‘台农一号’芒果香气物质[J]. 中国农业科学, 2025, 58(3): 564-581. |
| [13] | 王凡, 刘陈玮, 陆红臣, 徐仁超, 卞晓春. 蚕豆响应交链格孢侵染的转录组分析及VfPR4的抗病功能验证[J]. 中国农业科学, 2025, 58(22): 4656-4672. |
| [14] | 穆赢通, 路景诗, 张雨桐, 石凤翎. 基于转录组和WGCNA的直立型花苜蓿抗旱关键基因识别[J]. 中国农业科学, 2025, 58(21): 4528-4543. |
| [15] | 潘媛, 王德, 刘楠, 孟祥龙, 戴蓬博, 李波, 胡同乐, 王树桐, 曹克强, 王亚南. 两种高通量测序技术鉴定苹果病毒效果评价及两种新病毒的鉴定[J]. 中国农业科学, 2025, 58(2): 266-280. |
|
||